
医谷编者案:这是小编偶尔发现的偏长诗,小编自叹弗如。这篇《关于药品注册管理》的长诗发表于大概2008年前后,虽然时间有点远,可能政策也有所变动,但对了解和掌握药品注册非常有帮助,特推荐之。
(1)都说药品有三性,安全有效质可控;根据两法一条例,注册管理办法定。
(2)临床生产须申请,进口检验审批中;注册相关之监管,正是办法活动圈。
(3)所谓药品之注册,就是申请被许可;国局依法审查后,决定批准或者否。
(4)国家鼓励搞创新,审批通道有不同;专治疑难危重症,若是新药可特审。
(5)临床上市谁许可,谁是注册老大哥;国局统抓总负责,审批大权手中握。
(6)三公原则真不错,关键还得看效果;注册制度已确立,坚定不移抓落实。主审集体负责制,相关公示回避制;受检审批各环节,加强监管不松懈。
(7)药品注册审批中,重大事项需慎重;涉及公众利益时,发布公告且听证。许可涉及大利害,当局作出决定前,告知利害相关人,有权听证和申辩。
(8)受检审评结论等,当局指路供查询,注册时限和流程,申请目录和范本。药品注册每一环,相关人员有名单。药品基础数据库,可查批准药目录
(9)申报资料有数据,数据背后是技术,药监药检相关人,负责保密不透风。
(10)药品注册申请人,提出申请担责任;合法登记独担责,境内申请有资格。谁做境外申请人,非是药企无身份;办理进口药注册,境内机构可受托。
(11)常见申请有五个,新仿进补再注册;境内申请走新仿,境外进口不一样。
(12)境内未曾上市药,申请需按新药报;改剂改给新增症,貌似新药走流程。仿制已有国标准,仿生制品依然新;境外申请进口药,欲在境内上市销。改变原来批准项,补充申请来帮忙;药品批文效期满,再来注册续生产。
(13)研究数据须充分,证明药品有三性;药物研究当真做,申请之人须负责。
(14)注册资料引文献,应当注明其来源;参考文献列出来,著作刊名期页卷。文献资料未公开,提供证明性文件;外文资料按规定,依照要求附中文。
(15)行业发展应有序,药品市值可评估;产业发展有规划,上市药品再评价。
(16)药品注册申报中,药监核查在进行;现场有因产现检,确认资料真准整。
(17)两位以上申请人,药企所在地申请;都是药品生产者,制剂当局应负责。倘若都是研究所,试制现场有话说。受理管辖并不难,样品试制很关键。
(18)药品处方工艺等,专利权属须说明;国内专利有他人,提供不侵权声明。申请说明和声明,网站公示可查询;专利纠纷有发生,国局不是当事人。
(19)他人已获专利权,可报届满两年前;国局审查合规定,专利期后发批文。
(20)销售生产NCE,相关数据要保密,许可之后六年里,非法数据难获批。
(21)药物临床前研究,内容杂应有尽有。合成提取不能少,理化性质纯度高。剂型处方要筛选,工艺方法经检验;质量指标稳定性,药理毒理分量重。中药制剂原药材,加工炮制和来源;生物制品起始物,菌毒种和细胞株。材料来源和标准,适宜条件下保存;遗传稳定及特征,免疫研究在当中。
(22)临床前研究过程,遵循相关之规定;其中评价安全性,执行GLP规定。
(23)研究机构软硬件,在研品种相关联;动物试剂原材料,符合要求很重要。
(24)委托试制或研究,委托合同必须有;注册申报要说明,担责不在受托人。
(25)药物制剂独申请,原料药需有批文;当然注册证也行,合法获得来路明。如果无文也无证,需经国家局批准。原料制剂关系亲,选择来源当慎重。
(26)境外研究也可行,项目页码要说明;公证证明不可缺,必要核查防出错。
(27)药监感觉有疑问,可以要求复验证;可托药品检验所,研究机构也能做。
(28)指导原则可参考,药物研究弯路少;其他评价和技术,证明资料说清楚。
(29)药品批文已拿下,核准工艺是框架。监督检查何所凭,批准工艺和标准。
(30)药物临床含等效,国家批准很必要;临床执行GCP,监督检查更有利。
(31)注册申请报新药,临床验证不能少;仿制补充做或免,具体要求见附件。临床细分有四期,一二三期后上市。I期主评安全性,药理药代也进行。II期初步判疗效,安全未必不重要;随机盲法加对照,III期方案要参考。III期临床是确证,再评疗效安全性;看看风险效益比,关系是否能获批。III期临床需上量,随机对照还设盲。IV期上市后进行,关注疗效和反应。说说生物等效性,参比试剂有异同;比较动力学参数,统计结果得结论。
(32)临床受试病例数,符合要求是正路;本法后面有附件,特殊情况可减免。减免临床申报提,过后恐怕很难批。II期III期数百例,等效也就二十几。
(33)毒种选种制疫苗,或者其他特殊药;确无动物评其效,缺少药理也能报。只要安全有保证,人体试验可申请。此种情况是例外,适用范围比较窄。
(34)临床批件拿到后,临床试验可动手;试验机构谨慎择,经认定后有资格。
(35)临床试验所用药,GMP车间造;样品制备可委托,申请之人负总责。
(36)临床样品必须检,申报标准自检验;也可委托药检所,总之药品必合格。血液制品和疫苗,生物制品风险高;国局指定药检所,药监抽验不能少。
(37)临床试验实施前,备案资料到药监;方案单位研究者,知情伦理不能缺。
(38)申请人有监察员,临床机构莫乱来;违规未按方案做,有权督促其改错。
违规情况很严重,试验终止或暂停;同时上报药监局,而后谋划下一步。
(39)药物临床试验罢,资料报告交国家;同时还交数据库,且看疗效实还虚。
(40)临批到手三年后,不做临床命自休;他日意欲继续做,重新申报等结果。
(41)临床试验过程中,严重不良事发生;研究人员快行动,在一天内告多人。国家地方药监局,申请人也要清楚;及时报告伦理委,点背不能怨社会。
(42)下列情形若有一,国局当然很生气;命申请人改方案,暂停终止很难堪。
伦理职责未履行,受试安全无保证,严重不良延期报,证实药物没有效。
试药质量有问题,弄虚作假不客气,其他有违GCP,药监一查查到底。
临床试验过程中,及时督察结果真,莫要等到全剧终,很多事已说不清。
(43)不良反应非预期,而且出现大面积,或者严重不良事,药物质量大问题,
药监部门紧急控,以防事态更严重,责令试验终暂停,相关单位要执行。
(44)老外缺乏样本量,欲在国内做临床,所谓国际多中心,应向国局提申请。
试药已在外注册,IIIII期药也不错。未注册预防疫苗,国局大声说不要。
批准国际多中心,I期可以复进行。国际多中试验中,信息共享要充分。
出现重不良反应,无论哪国有发生,包括非预期不良,老外都要告我方。
临床试验已做毕,试验资料需整理;试验报告当完整,而后再往国局送。
国际多中心临床,国内注册欲分享,符合临床之要求,提供研究之所有。
(45)特批办法另行定,下列申请可特审:一般是真一类药,中二也走该通道。
艾滋癌症罕见病,这些新药优势明,有病无药可治疗,可走特批之通道。
申请单位提申请,药审专家与会论;决定那些搞“特殊”,防止滥竽也充数。
(46)多个单位研制药,只许一位去申报;不得重复去申请,联合申请共署名。
尽管申请人数众,但是批文属一人。即使同品不同规,不能分属俩单位。
(47)改剂但不改途径,采用技术应当新,提高质量安全性,临床优势要说明。
改剂但不改途径,或者新增适应症,必由药厂提申请,条件不备也不行。
靶向制剂缓控释,研究起来有难度;因此不必是药企,科研院所可以提。
(48)新药审批过程中,分类要求不变更,即使国内外上市,即使成分也相同。
(49)注册资料一次性,更新资料勿补充;特批申请是例外,更有安全新发现。
通常如果需要补,申请撤回是条路;重新申报需关注,法规监测期有无。
(50)新药完成临床前,注册申请表要填;资料和表报省局,可对送样说声“不”。
(51)省局大概看一看,合要求出受理单;不合要求说原因,通知不受该申请。
(52)受理之后5日内,省局组建核查队;查看现场和记录,资料审查是初步。
审查结束出意见,生物制品有特点;仨批样品需抽验,告药检所赶快检。
(53)审查意见和资料,还有核查后报告,限期药审中心交,申请单位通知到。
(54)药检接到通知后,检验工作始动手,依据生物药标准,判定是否合格品。
检验固然很重要,标准复核不能少,报告送交CDE,抄送申请人那里。
(55)药审中心资料到,组织专家审资料,审评专家来源广,审评时间莫太长。
有时需要补资料,说明理由很重要,技术审评一结束,意见资料到国局。
依据审评之意见,国局权力得实现,符合规定准试验,临床试验可开展。
不合则获通知件,申请单位很失败,国局不批有理由,数年辛苦付东流。
(56)临床做完生产报,依然填写申请表;报产资料送省局,上市申请已上路。
若有自制标准品,情况稍微有不同;有关资料原材料,中检所里等你报。
(57)省局形式再审查,符合要求受理它;不符规定不受理,要求补正或补齐。
(58)受理五日去核查,检查临床真还假;试制现场当然看,资料初审是一环。
所有环节结束后,审查意见应该有。样品需抽三批号,这里不含生物药。
样品送往药检所,药品标准待复核。意见报告和资料,限期向国局送交。
申请单位需通知,不能蒙其在鼓里。这些环节审评前,更大考验在后面。
(59)药检复核药标准,复核时限有规定,意见送往药审中,抄送局里申请人。
(60) CDE收资料后,医药专家显身手;资料审评限期完,需补资料说理由。
符合审评之规定,马上告诉申请人,申请药品产现检,认证中心有关联。
不符规定很抱歉,往国家局转意见;国局据此作决定,理所当然不批准。
发给审批通知件,退审理由很冠冕。申请单位很受伤,上市愿望泡成汤。
(61)接到通知半年内,申请单位做准备,现检申请递哪里,牧童遥指CCD。
(62)认证中心收申请,三十天内要行动,确认工艺可行性,动态抽样令人惊。
非生物药一批样,生物药抽三批量,复核药检所里送,报告十日到药审。
(63)认证车间生产样,少有余地可商量。新企新车增剂型,合乎规范生产中。
(64)药检收到抽检品,限期检验依标准;报告送交CDE,抄送单位和局里。
(65)检查检验和审评,药审中心做汇总;形成综合之意见,依法转到局里面。
国局根据该意见,是生是死局长签;合规定发新药证,具备条件发批文。
不符规定很无奈,收获审批通知件;辛辛苦苦好几年,转眼回到临床前。
改剂不改变途径,或者新增适应症,批准后无新药证,除了靶向缓控等。
(66)公众健康要保护,新药安全需关注;国家设立监测期,最长上市五年毕。
新药监测期期间,药品三性需再验;他人上市或改剂,国家一律不给批。
(67)新药监测期限内,药企应有所作为;考察工艺和质量,疗效安全怎么样?
每年都向省局报,第一负责人做好。药企未履行责任,省局责令其改正。
(68)相关单位有发现,新药质劣不安全,及时向省局报告,省局调查并上报。
(69)新药设立监测期,获准生产两年里,如若生产没进行,同类申请可获准。
后批新药上市后,上市监测又从头,这一条款实无用,数年难遇一情形。
(70)新药进入监测期,有的临床已经批,可以按部就班做,进口药可免监测。
(71)新药进入监测期,同品申请不受理;如果没有批临床,出师未捷身先亡。
进口品种不例外,退审感觉很失败。待到监测期满后,申请仿制或进口。
(72)进口申请抢了先,率先上市放光彩,境内有人批临床,可撤亦可继续上。
撤回可以提仿制,继续可申请上市。临床批件没到手,退回再按仿制走。
(73)仿制当然是药企,申请之时需注意,生产许可范围内,是否与药相匹配。
(74)仿制并非很容易,与被仿药关系密,治疗成分当相同,相同规格同剂型。
给药途径不许动,治疗作用应相等。若有多家生产商,对照研究要加强。
(75)注册申请仿制药,填写注册申请表,现检申请和资料,一起递至省局报。
(76)申报资料到省局,形式审查头一步,合乎要求则受理,反之说“不”讲道理。
中保受理之日起,意欲仿制是难题,不予保护可仿制,给予中保七年期。
(77)受理程序差不多,五天之内去考核,核查研制软硬件,同时进行产现检。
现场检查过程中,抽取三批待检品,送往药品检验所,一俩月内出结果。
新药核查有先后,仿制核查同步走,注册核查不简单,样品生产要规范。
(78)申报资料需审查,审查意见省局拿,符合规定向上交,意见报告加资料。
这些东东寄药审,同时通知申请人;不合规定很无奈,发给审批通知件。
理由当然要说明,通知药检检验停。未上战场先阵亡,英雄焉能不感伤。
(79)药检检验抽检品,注意时限有规定;报告仍交CDE,抄送单位和局里。
(80)收到资料和意见,审评工作始开展,法律规定有时限,审评才是真考验。
如有必要会发补,申请单位莫叫苦,补充资料留希望,至少总比退审强。
(81)检查检验报告到,技术审评更重要,三部汇成总意见,转战国局化批件。
合乎规定发批文,或补临床再验证,不符规定通知件,心中有怨也无言。
(82)临床试验结束后,资料再向药审走,国局最终做决定,发通知件或批文。
(83)上市药品有问题,安全风险已无疑,国局暂停其受理,在审品种停审批。
(84) 注册申请进口药,境外上市很必要;异地上市未许可,老大点头开先河。
进口注册药生产,过程管理须规范;符合双GMP,境内以及所在地。
(85) 注册填写申请表,报送样品和资料;提供相关之证明,向国家局提申请。
(86) 形式审查不可少,没有大碍很重要;符合要求则受理,告中检所验三批。
不合要求不受理,老外得知很生气;老大说明个中由,老外听了直点头。
国局认为有必要,出国检查不蹊跷;检查产研之现场,当场抽取若干样。
(87) 检品来到中检所,资料标准待复核;规定时限是五天,组织人员开始检。
(88) 承担进口药检所,资样对照先收着,限六十天检验完,报告中检所里传。
特殊药品和疫苗,短期难以出报告,复核时限有所延,总计不过九十天。
(89) 进口标准已复核,报告亦抵中检所;二十天内找专家,进行技术再审查。
进口检验很麻烦,过了一关又一关;审查意见出结果,偶有标准再复核。
(90) 注册检验终完成,报告复核后标准,还有意见之复核,依依惜别中检所。
这些来到CDE,审评按钮已开启,同时抄送申请人,申请单位应知情。
(91) 药审中心开审评,医药专家显神通,必要要求补资料,补充理由要说到。
(92) 审评意见很关键,检验结果非等闲,综合形成总结果,审评暂告一段落。
国局依据总意见,审批大权威力现,合规发给临批件,不符则获通知件。
不予批准讲理由,君子动口不动手。注册之路弯又弯,非是按部或就班。
(93) 临床试验获批准,要求没有大不同;参见本法第三章,依照要求做临床。
临床试验结束了,应当再填申请表;报送资料依规定,临床变更和补充。
报资料时需说明,依据理由当充分,提供证明性文件,宁多勿缺较保险。
(94) 药审专家再会战,重点在临床试验;依然可能补资料,说明理由很重要。
所有意见再汇总,国局据此做决定。合规发给注册证,不符则不予批准。
外国曰进口药品,台港澳地稍不同。不批令人很吃惊,具体理由需说明。
(95) 申请进口药制剂,注意内包和容器,还有所用原辅料,合法证明需提交。
原辅国内未批准,用于制剂有担心,相关资料当提供,工艺质量标准等。
(96) 进口药品分包装,好比大箱变小箱,制剂生产境外完,转战国内包装换。
内包药品搞外包,外包如今很新潮,勿忘放置说明书,贴上标签闯江湖。
(97) 申请进口药分装,非任何药都能上。无规难以成方圆,分包注册有条件。
先获进口注册证,医药注册证也行。其次国产药没有,或者有供不应求。
第三一夫一妻制,同厂同品不二企。分包装期有限制,不超注册证效期。
第四除了片胶囊,境外做完内包装,接受分包之药企,生产许可毫无疑。
若是裸片或胶囊,药企还要内包装,GMP认证范围,当与剂型相匹配。
申请进口药分装,把握时机路宽广,证期届满一年前,更晚申请更加悬。
(98) 境外厂商境内企,分包合同定关系;当填补充申请表,批准之后可分包。
(99) 申请进口药分装,药企提出没商量,资料样品哪里去,径直所在地省局。
这里有条需强调,老外填写申请表。省局形式审查后,受理表明合要求。
不合规定不受理,阐明原因讲道理,境外厂商很失意,境内药企很生气。
审核意见省局提,连同资料报国批,同时通知申请人,精心等待候批文。
(100) 国局当然审资料,合规定则发文号,不符规定通知件,失败多了自坦然。
(101) 进口分装之药品,执行进口药标准。原装分装一家人,标准自然无不同。
(102) 分装药品说明书,与外一致不冲突,标签一致不多语,批文企业要标注。
(103) 境外大包制剂检,依照规定去化验,进口分装后产品,执行检验用标准。
(104) 供药境外制药厂,负责药品之质量,分包药品出问题,撤销文号不客气。
必要撤销注册证,看来问题很严重,药品管理法规定,谁干坏事受严惩。
(105) 仿制药属非处方,申请表标附加项,申报资料需说明,符合要求可批准。
(106) 仿制药是双跨品,谨慎择一提申请;不可兼得鱼熊掌,双跨品种也一样。
(107) 非仿申报非处方,表中亦标附加项,有些新药或改剂,可按非处方审批。
相关规定都符合,方有可能获许可;不符规定不予批,按处方药批管理。
非处方药改剂型,但不改变适应症,途径不改剂量对,有望申请OTC。
另外还有新复方,非处组方有希望。至于如何做临床,本法似乎没有讲。
(108) 注册申请OTC,五六资料当遵规,除了二十四号令,还有非处方规定。
(109) 进口申请OTC,申报审批按常规,技术要求同国内,身体健康是最美。
(110) 药品已然获批文,有些事项欲变更,或改附件中东东,都属于补充申请。
变更事项莫乱来,切忌臆断拍脑袋;技术原则指引下,变更影响要评价。
评估重点是三性,研究工作可作证。若不产生大影响,或许可以免临床。
(111) 补充申请也填表,当地省局收资料;形式审查省局搞,流程还是老一套。
符合要求受理之,不合规定不受理,当然理由要说明,申请人应仔细听。
(112) 进口药品补申请,国局报料加说明;境外监管也要批,批准文件交局里。
国家局形式审查,其实是走马观花,受理与否看要求,不予受理讲理由。
(113) 修改检验用标准,药用辅料欲变更,影响质量改工艺,省局提议国局批。
审核意见报送前,申请人有知情权。修改标准补申请,省所或复核标准。
(114) 国内企业名称变,国产药品效期改,改变厂内生产地,都是省局受理批。
合规发放补批件,备案国局管理便;不符规定通知件,此种情形是意外。
(115) 包装标签依规变,说明书按要求改,这类申请更简单,请到省局去备案。(116) 进口药品补申请,统统都由国局审。有些申请简单办,只需国家局备案。
改变原料药来源,标准不改外观变,说明书依法修订,包装标签变更等。
(117) 生产技术之转让,变更影响药质量;省局出马产现检,现抽三批送检验。
(118) 国局审查补申请,资料可能会补充。补充资料不用怕,认认真真应对它。
合规发给补批件,不符收获通知件;不批当然有理由,遭遇退审难回头。
(119) 补充申请批准后,换新批文注销旧;增发药品批文号,原批文件仍有效。
(120) 药品批文效五年,效期届满欲续延,失效之前六个月,赶快申请再注册
(121) 批文有效期限内,申请之人应有为,关注药品之三性,相关结果系统评。
(122) 药品批文持有人,面向省局提申请,填写再注申请表,申报资料不能少。
进口再注册申请,国家局受理大厅,申报资料申请表,哪一个也不能少。
(123) 省局依然查资料,套路还是那几招,合乎要求则受理,不符要求则拒之。
(124) 受理之后六个月,正审完毕再注册,符合规定许再注,反之上报国家局。
(125) 国审进口再注册,时限依然六个月;符合规定让通过,反之不予再注册。
不许再注说理由,老外听后直摇头,话说三遍淡如水,一腔热情终成灰。
(126) 有药不符合规定,再注当然不批准。再注册也有条件,下列药品在圈外:
效期满后提申请,无视办法之规定;生产上市过程中,国局要求耳旁风。
国家经过再评价,效差不良反应大,其他危害人健康,申请再注没希望。
批准证明被撤销,生产条件很不好;监测期内未履责,其他情形不好说。
(127) 省局意见到国家,国局组织再审查,不符规定不再注,理由应当说清楚。
药未通过再注册,期满批文就没了。五年弹指一挥间,批文转眼化云烟。
(128) 注册检验有两种,检样和复核标准。检验依据是标准,判定是否合格品。
药品标准之复核,主体当然药检所,重点在于方法学,既要检验更审核。
方法可行且科学,指标项目合理设。药品注册检验完,报告意见往上传。
(129) 注册检验承担者,省所或者中检所;进口药品需检验,组织实施是中检。
(130) 有些药品很重要,检验单位国家找;一般都在中检所,或者指定承担者。
特批品种前两条,生物制品放射药;另外还有其他药,指定药检所报到。
(131) 特批药品有特权,检验复核应优先。政策不停在扶持,鲜有新药有价值
(132) 药检所里做检验,具备一定软硬件;实验必须规范做,计量认证应合格。
人员检验水平高,设备仪器性能好;检验质量有保证,分析结果无疑问。
(133) 单位送样和资料,配合抽样供对照;报送抽取多少样,全检用量三倍量。
生物制品更特殊,注册检验要记录;检定记录批对应,切莫以假去乱真。
(134) 复核新药之标准,样品检验在其中,根据数据和标准,依规方法学验证。
标准复核一结束,复核意见便出具;标准项目合理否,意见终往国家走。
(135) 要求重新订标准,复核单位靠边请;纵然申请人委托,原药检所当拒绝。
(136) 国标当是局颁布,中国药典较特殊;注册及其他药品,都在国标体系中。
标准旨在控质量,必有质量指标项,检验方法不用说,制法偶尔搭上车。
药品注册之标准,国局批给申请人;生产药企须执行,不低于药典标准。
(137) 标准项目方法定,可在药典取正经,看看凡例和附录,相关原则要清楚。
(138) 标准研究不容易,代表样品是前提;稳定工艺和质量,制得一定规模样。
(139) 标准物质有四类,分析校验常需备;标准品和参考品,对照药材对照品。
特性量值已确定,专供标准测试用,设备校准方法评,供试药品值待定。
(140) 标物标定谁负责,理所当然中检所;省所企业药检所,负有义务协助做。
(141) 负责标定中检所,全部资料皆审核:标物原材料选择、标定方法和结果、制法定值准确性、量值溯源可稳定、分装包装条件等,据此做出终结论。
(142) 品名标签说明书,遵规守法不逾矩;依照原则命药品,五六二十四号令。
(143) 药品标签说明书,注册申请人提出;药审中心先审核,国局批产时负责。
说明标签讲科学,不仅规范且准确,切忌误导医患者,出错企业应担责。
(144) 药品生产上市后,申请单位非无忧,跟踪安全有效性,补充申请修说明。
(145) 印制说明书标签,不符要求有危险;按照国家格式来,核准内容不宜改。
(146) 药监部门亦守法,法规时限非空话;注册时限是何意,受审查批最长期。
中止审批不计时,补充资料时停滞。注册检验和审评,执行本法之规定。
特殊原因需顺延,说明理由情可原;上报国家局批准,谁会告知申请人?
(147) 药监接到申请后,形查决定受不受。依法不需要许可,不受理也不是错。
依法不在权限内,不予受理做得对,但是不宜拂袖去,应当为人指明路。
申报资料有错误,可以现更当允许。药监药企本一家,有严有爱方融洽。
形式不符不齐全,一次告知限五天;补正逾期不告知,收资料日即受理。
部门职权范围内,资料齐全合法规,或按要求已补正,应当受理该申请。
不论受理不受理,通知盖章有日期,注册专用章一盖,形式审查不再来。
(148) 受理之后三十天,省局不能太清闲;初审资料现场核,抽样通知药检所。
审查意见核报告,连同申报之资料,一并报送至北京,意见通知申请人。
(149) 注册检验有时限,纯检一般三十天;检验之外还复核,时限延至两个月。
特殊药品和疫苗,安全风险级别高;待到他日出结果,更比常规多一月。
临床用药抽查检,多是药检所检验,按照前款定时限,限期检完不宜延。
(150) 审评工作是重点,重点当然有时限。新药临床九十天,特批品种十日减。
新药生产一百五,特批少吃一月苦;仿改一百六十天,审评似乎会更严。
补充申请需审评,四十天后离中心;注册申请进口药,审评时限可参照。
(151) 审评过程需下补,应一次性发出去;申请人若有异议,意见可以当面提。
补充时限四个月,一次补齐没的说;发补品种走特批,按照要求去办理。
发补之后续审评,审评过程近尾声;原时限内三有一,四分之一是特批。
药品注册申请人,突发奇想撤申请,审批程序自然停,从此无人再问津。
(152) 国局审批二十天,领导批准延时限;延长十日是为何,告申请人解疑惑。
(153) 国局决定审批后,再过十天证到手。苦盼数年等这天,但愿不是通知件。
(154) 下列情形若有一,当局当然不予批:无缘无故相雷同,资料不真难证明。
项目设计和实施,药品三性不支持;显示三性有缺陷,上补超时无悬念。
原料来源不合规,现场核查惹是非;样品检验不合格,其他情形不好说。
(155) 不受不批说理由,申请单位不好受;虽可复议或诉讼,不可沽名学昆明。
(156) 国局不批有异议,申请复审俩月里;填写复审申请表,说明理由报材料。
复审内容有局限,原报资料事项圈;暗示复审希望小,申请复审准备好。
(157) 复审申请到国局,决定五十天作出;维持原判不批准,已不允许再复审。
(158) 复审需要再审评,资料重回药审中;所谓参照原时限,莫非又要从头来?
(159) 行政许可法规定,胡作非为可不行,即使骗得批文到,国局依法可撤销。
(160) 药监人员违法规,若有下列之行为:轻则批评其改正,重则依法给处分:
符合条件不受理,材料依法不公示,受审批评过程中,当告不告申请人。
资料不符或不齐,告知补正非一次;不受不批理不明,依法听证不听证。
(161) 药监人员工作中,两袖清风为官正;科学监管为谁管,心态不正前程断。
索取收受钱财物,不当得利毁仕途。构成犯罪受刑罚,轻则行政处分他。
(162) 法规貌似很严厉,实施过程却无力,下列情形稍严重,有责有处或上刑:
不符合条件申请,批准或却越权批准;法定条件已符合,不批或延期注册。
保密义务未履行,技术数据泄他人。当官不为民服务,不如回家卖红薯。
(163) 药检出具假报告,触犯刑法罪难逃;轻则警告给处分,造成损失担责任。
(164) 擅自收费乱收费,上级领导责成退;依法处分相关人,不正之风刮不停。
(165) 违反GCLP,依法处罚当严厉,警告罚款停业整,失去资格亦可能。
(166) 申请单位报临床,资料不真送假样,老大发现很生气,手中权力显威力。
不予受理不批准,严肃警告申请人;申请人再报临床,一年之内是妄想。
已批临床撤批件,罚款一至三万元,该项申请欲再来,至少还要等三年。
不良行为应记录,药监部门当公布;法规实施一年半,未有人上黑名单。
(167) 报产进口药申请,资料有假样不真,不受不批还警告,一年不收其资料。
生产进口已批准,药监撤销其批文;五年不受理申请,一至三万元罚金。
(168) 要求试验再重复,竟然有人敢说不,国局警告令改正,顽抗到底不批准。
(169) 有下列之一情形,国局注销其批文:期满申请人自销,再注申请肥皂泡;
生产许可证吊缴,药毒批文被撤销;行政处罚撤批文,其他撤批文情形。
(170) 中天化生补再注,附件一二三四五;首现新药监测期,最后一个附件里。
(171) 批文格式有规定,国药准字打头阵;化中生进分拼音,后四位年四位顺。
进口医药注册证,格式稍微有不同,汉语拼音第一名,后八依然年和顺。
境内分包大包证,原证号由B引领;新药证书更简单,准字换“证”即编完。
(172) 注册申请省受理,补充再注册审批,尽管是由省局做,其实受国局委托。
如果国局再放权,省局能力尽施展,注册审评和审批,何时下放不定期。
(173) 上市药品要编码,标签工作量加大;编码规定另行定,法规出台太匆匆。
(174) 麻精毒放特殊药,注册管理把关好,不仅符合本法规,相关规定不违背。
(175) 批文管理中药材,进口药材和饮片;如何管理打补丁,补丁至今未现身。
(176) 技术转让产委托,新衣补丁真是多;征求一遍又一遍,千呼万唤不露面。
(177) 二零零七年国庆,恰逢新法正施行,原局第十七号令,当天完成其使命。
年年岁岁法相近,岁岁年年人不同,法若有情法亦老,政策稳定是正道。
28号令十五章
总则要求做临床,新药研罢仿制上,进口药品非处方,补再注册检验章。
标准说明时限长,复审无情法有网,附则补丁真是多,新法出台好匆忙。
来源:互联网

为你推荐
 资讯
资讯 国家药监局批准两款创新医疗器械
近日,国家药监局批准两款创新医疗器械上市。分别为苏州无双医疗设备有限公司的植入式心律转复除颤器和深圳大医伽玛刀科技有限公司的头部伽玛射线立体定向放射治疗系统。
2026-07-15 18:34
 资讯
资讯 翰森制药最高23亿美元授权并参与纳斯达克上市公司股权投资
7月14日,翰森制药发布公告宣布,已与Avere Therapeutics, Inc (Avere)签订独家许可协议及参与战略投资。
2026-07-15 14:20
 资讯
资讯 2026年底前至少完成一轮集中采购,国家卫生健康委 国家中医药管理局 国家疾病预防控制局关于进一步做好公立医疗卫生机构医用设备集中采购工作
国家级集中采购。国家卫生健康委继续负责组织全国公立医疗卫生机构甲类大型医用设备集中采购。个性化定制程度高、需配套设计并建设基础设施工程、逐台按程序获得注册的设备,待...
2026-07-15 09:27
 资讯
资讯 中国医药工业主营业务收入100强
近日,由中国医药工业信息中心举办的2026第43届全国医药工业信息年会在北京开幕,备受关注的“2025年度中国医药工业主营业务收入前100位企业”同步揭晓。
2026-07-14 19:55
 资讯
资讯 国家药监局批准1类创新药阿更葡糖钠注射液上市
近日,国家药品监督管理局批准杭州奥默医药股份有限公司申报的1类创新药阿更葡糖钠注射液(商品名称:奥美克松)上市,该药品用于拮抗罗库溴铵诱导的神经肌肉阻滞。该药品上市为...
2026-07-14 17:01
 资讯
资讯 迪哲医药最高15亿美元BD阿斯利康
今日,迪哲医药发布公告,公司授予阿斯利康在全球范围内的独家开发、商业化舒沃哲的权利。公司将获得阿斯利康支付的一次性、不可返还的首付款6亿美元,最高达4亿美元的临床开发...
2026-07-14 14:57
 资讯
资讯 国家医保局、国家药监局开展跨部门联合检查
近日,国家医保局、国家药监局组成联合检查组,依据药品追溯码筛查线索对内蒙古自治区和山西省部分地市医药机构、药品批发企业开展联合检查。国家医保局党组成员、副局长黄华...
2026-07-14 11:46
 资讯
资讯 2026年国家医保目录及商保目录调整通过形式审查名单公布
2026年目录调整申报阶段,国家医保局共收到基本医保目录申报信息800份,涉及药品通用名664个,最终601个通过形式审查,其中目录外368个,目录内233个,总体通过率91%。共收到商...
2026-07-14 10:34


 资讯
资讯 全国首笔药品追溯码无追索权保理业务在温州落地
近日,在温州市医保局的积极推动下,全国首笔基于药品追溯码的无追索权保理业务成功落地。国药控股温州有限公司凭借药品追溯码形成的真实交易记录,将其对温州医科大学附属眼视...
2026-07-13 10:15

 资讯
资讯 时隔8年调整的2026版国家基药目录新增的4种1类创新药和独家品种
7月9日,国家卫生健康委、国家中医药局、国家疾控局三部门公布2026年版国家基本药物目录。国家基本药物目录在新医改启动后,于2009、2012、2018年完成三轮更新,这是时隔8年后的...
2026-07-12 21:31
 资讯
资讯 陈薇院士出任中国工程院党组成员、副院长
据中国工程院官网消息,根据中共中央和国务院通知,中国工程院第九届院领导班子组成如下:张玉卓同志任党组书记、院长;张军、陈杰、李仲平、陈建峰、陈薇等同志任党组成员、副...
2026-07-11 21:36

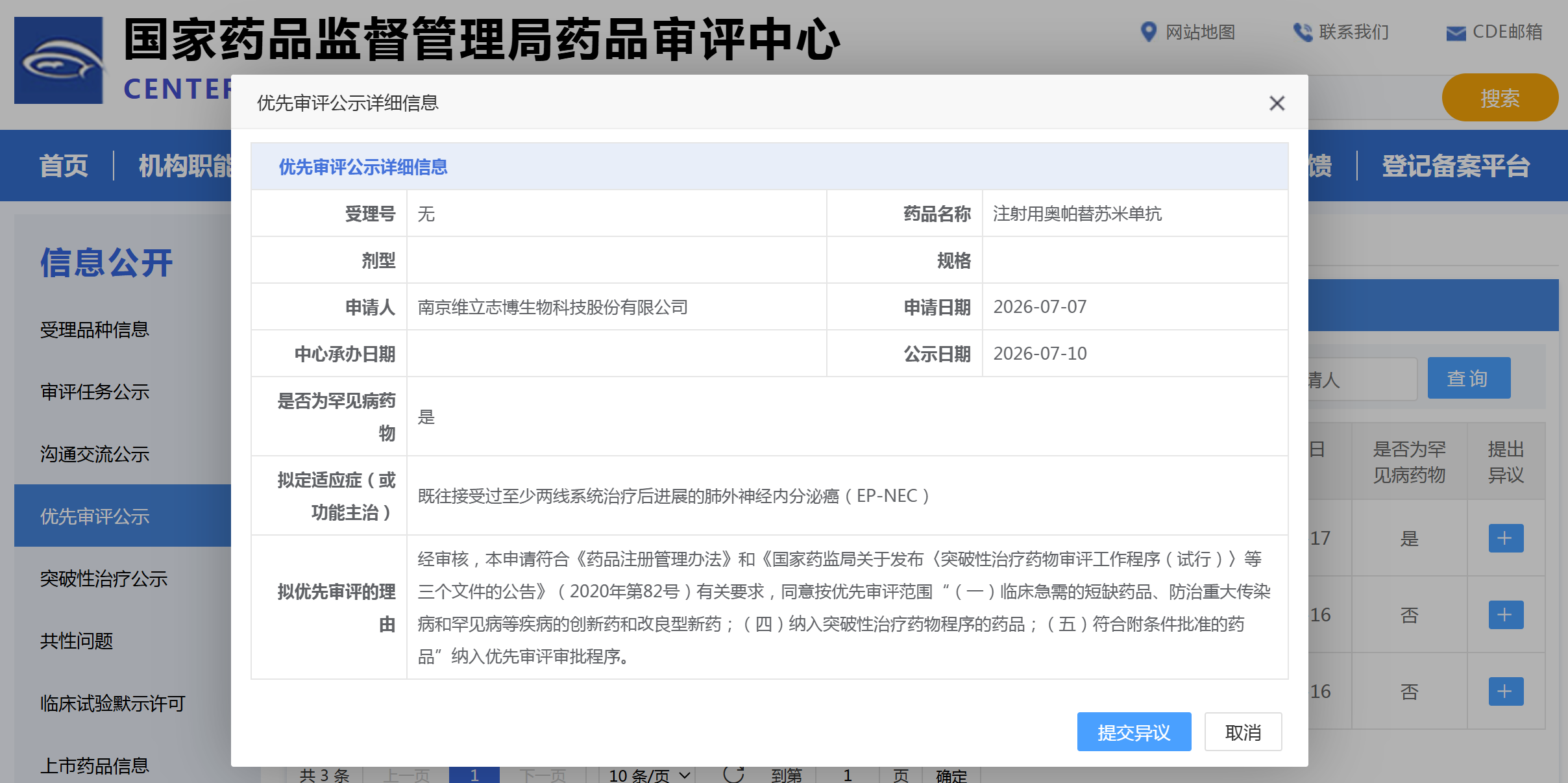 资讯
资讯 维立志博PD-L1/4-1BB 双抗拟纳入优先审评
7月10日,国家药监局药品审评中心官网显示,南京维立志博生物自主研发的注射用奥帕替苏米单抗拟纳入优先审评品种,申报适应症为既往接受过至少两线系统治疗后进展的肺外神经内分...
2026-07-10 15:08
 资讯
资讯 丰原药业实控人拟变更为安徽蚌埠市国资委
7月9日,丰原药业发布公告,公司控股股东安徽丰原集团有限公司的一致行动人7月8日与蚌埠投资集团有限公司签署《股份转让意向协议》,蚌埠投资集团拟受让丰原集团的一致行动人持...
2026-07-10 13:49