
系列1. 生物制品中残留DNA检测标准的变化
2015年3月底,中国食品药品检定研究院网站的国家药品标准物质目录中新增加了一个编号为410001的产品:CHO宿主细胞DNA残留检测试剂盒(PCR-荧光探针法)。这个由中检院与中科院联合研发,由生物制药企业参与标定的试剂盒与现行美国药典(USP)建议的检测方法同步,但与2010版药典附录中外源性 DNA 残留量测定法有所不同,可能会对国内生物制品的研发和生产的上下游企业产生影响。
生物制品中宿主细胞残留DNA具有潜在致瘤和传染风险,所以各国药品监管部门对DNA杂质的限量要求非常严格。美国药典在General Chapter <1130>介绍了三种常用技术,但将在2015年颁布的新版(USP38-NF33)中增加全新章节(General Chapter <30>)来进一步规范残留DNA检测的方法和标准物质。与1000号以上的章节不同的是,USP编号1000以内的章节详细规定了检测技术、系统适应性标准和标准物质。新版USP中将唯一推荐qPCR法作为生物制品中宿主残留DNA的标准方法。qPCR法的技术优势在于序列特异性高、灵敏度高、重现性好,可以为生物制药工业在工艺研究和成品质量控制方面提供可靠的检测手段。
我国参照WHO、FDA和欧盟的标准,很早以前开始就对生物制品中残余DNA含量进行限制。从卫生部颁布的《人用重组DNA制品质量控制要点》到近年的《中国生物制品规程》都对DNA含量做了严格要求,部分标准高于国际标准。2010年版中国药典附录收录了DAN探针杂交法和荧光染料法,这两种方法都存在技术缺陷,很难达到杂质限量检测的灵敏度,已经被欧美药典摒弃。目前,仍有很多国内企业沿用这两种方法检测残留DNA,致使生产工艺和产品质量很难达到国际一流水平。根据残余DNA检测技术的发展趋势,小编预计我国2015版药典或增补版本中将出现qPCR方法。企业在生产与研发中采用中检院标准物质目录中提供的成套试剂盒,可以大幅度减少费时、耗力、成本高昂的方法学考察,只需开展少量方法适应性实验,就可以在生产工艺和质控体系的优化方面发挥实际作用,同时满足美国FDA相关标准的要求。同时,联合研发单位中科院湖州营养中心提供免费技术咨询和培训,帮助企业解决试剂盒应用中的各种技术问题。
生物制品可用于治疗和预防疾病,关系到患者和健康人的用药安全,产品质量必须得到保障。我国药品监管部门对生物制品中残留DNA的限量标准制订的非常严格,但药典修订存在一定滞后性,附录中的检测技术与先进国家尚存在差距,企业在研发和生产中应具有一定前瞻性,否则会使改进工艺、提高质量、保障安全的努力大打折扣。
系列2. 为什么要检测残余DNA?
生物制剂是制药行业中发展最快的领域,2014年全球十大畅销药中7个是生物制剂。这些销售上的重磅炸弹在临床上疗效确切,但研发成本高,生产和质量控制要求非常严格。绝大部分生物制剂是不经过胃肠道直接进入体内,所以除了生物活性外,监管部门对药品中杂质的限量要求非常严格。其中,宿主细胞残留DNA因为具有特别的潜在安全风险,一直是国内外药品监管机构关注的重点。美国药典会从2011年开始就组织专门小组讨论修订生物制品中残留DNA检测方法,并在2014年Prescription/Non-Prescription Stakeholder Forum Meeting#5上宣布将在2015年药典修订版中增加新的章节(General Chapter <30>)来规范检测方法和标准物质。为什么美国药典会的专家组花几年时间讨论一个微量成分(<100pg/剂量)的检测方法?还要专门增加章节来规范化?
回答这些问题,先要了解它的来源和潜在危害性。生物制品中的重组蛋白药、抗体药、疫苗等产品是用连续传代的动物细胞株表达生产,虽然经过严格的纯化工艺,但产品中仍有可能残余宿主细胞的DNA片段。这些残余DNA可能带来传染性或致瘤性风险,比如残留DNA可能携带HIV病毒或Ras癌基因。分布在哺乳动物细胞基因组的LINE-1序列可能发挥逆转录转座子作用插入到染色体中,这种插入可能影响关键基因功能的发挥,比如激活癌基因或抑制抑癌基因。
此外,由于微生物来源的基因组DNA富含CpG和非甲基化序列,增加了重组蛋白药物在体内的免疫源性风险。目前的研究结果显示,残留DNA的致瘤性相比传染性风险要低,但考虑到致瘤性实验是动物实验,传染性实验是在细胞水平做的,或许对两方面的风险都不能掉以轻心。众所周知,外源蛋白可能引起严重免疫反应,但关于残留DNA诱导的免疫反应的研究还不多。在一些临床前和临床研究中报道了高剂量的核酸样品,比如DNA疫苗或佐剂中的CpG寡聚核苷酸,可以诱导免疫反应,还诱导产生DNA抗体。
生物制品中宿主残余DNA既是是生产中带来的杂质,还存在一定安全隐患。因此,WHO和各国药物注册监管机构一般只允许生物制剂中存在100pg/剂量以下的残留DNA。根据杂质来源和工艺,特殊情况下最高允许10ng/剂量。
系列3. 各种残余核酸检测技术的优劣
正如前面讲过,2015年版的美国药典将新增章节对残余DNA检测进行规范化,那究竟和现有方法有什么不同?为什么最后只确定了一种方法?我们来看看现行美国药典对于残余DNA检测的总体要求[General Chapter〈1130〉 NUCLEIC ACID-BASED TECHNIQUES-APPROACHES FOR DETECTING TRACE NUCLEIC ACIDS (RESIDUAL DNA TESTING)]。“因为残留DNA涉及到潜在来源(传染性病毒DNA)、管理规程等关键问题,药品监管部门建议必须建立产品的DNA残留检测方法。不论是否成品的常规检测包含DNA残留含量检测,还是工艺开发中已经证实了DNA清除率,残留DNA技术指标和定量分析监测规程都必须确立。分析方法包括杂交法、基于DNA结合蛋白的免疫色谱法(阀值法)、定量PCR(Q-PCR)或其他DNA扩增方法。理想的定量检测方法的灵敏度应该能够检测到约10pg/剂量的残留DNA。杂交法、阀值法和定量PCR方法因为灵敏度可以达到检测要求,所以属于经典方法。”下面我们引用美国药典(USP)<1130>的内容分别介绍一下这三种方法。
杂交法(Hybridization-based):在这种方法中,根据宿主DNA序列设计DNA探针用于测定产品中配对DNA的数量。双链DNA被变性成单链后固定在尼龙膜或硝化纤维膜上,DNA探针被放射或荧光随机掺入标记以后,与膜上固定的样品宿主DNA杂交结合,并在胶片或成像仪对应位置中显现斑点。对于荧光标记的探针,斑点的光密度结果可以在仪器中定量分析。斑点光密度对应结合在目标DNA上的探针数,进而推测出残留DNA的数量。通过目测方法可以半定量地检测样品中残留DNA,仪器读片可以对应斑点光密度绘制标准曲线,对应检测结果更加准确。
DNA结合蛋白免疫阈值法(DNA-BINDING PROTEIN-BASED):这种方法使用DNA结合蛋白和DNA抗体,分四步检测。第一步,通过加热把DNA变性成单链DNA,变性后DNA与偶联了亲和素的DNA结合蛋白以及偶联了尿素酶的DNA单克隆抗体混合反应,液相中的单链DNA、DNA结合蛋白、DNA抗体共同形成序列非特异的复合物。第二步,样品混合液通过生物素标记的膜,亲和素-生物素结合把DNA复合物固定在膜上,洗去非特异吸附。第三步,膜放入检测仪器中与尿素溶液反应,反应产物氨改变溶液pH值并被仪器记录变化。这种pH值的变化直接与样品中的DNA数目相关。第四步,仪器软件自动分析原始数据确定样品中残留DNA数量。
定量PCR法(QUANTITATION PCR-BASED):qPCR方法以其快速、高通量的特点已经被应用于生物制药的一些领域(拷贝数检测与病毒检测)。这项技术能够确定各种样品中目标DNA序列的准确数量。DNA探针的设计非常关键,这种DNA探针包含一端染料分子和另一端淬灭分子。当特殊设计的DNA引物引导DNA聚合酶沿着模板序列复制合成另一条对应序列时,DNA聚合酶切断结合在目标DNA上的探针染料端,释放到反应液里的染料信号被仪器测量。经过数十个循环的DNA扩增,荧光信号与起始DNA模板成对应关系,对应标准曲线可以准确计算出样品中残留DNA的数量。
美国药典附录进一步对三种方法进行了应用评价。杂交法可以序列特异性地检测目标DNA,但32P标记的探针因为存在半衰期短、放射等问题,实际应用并不广泛。荧光标记的探针如果采用仪器读取信号,杂交法理论上可以达到定量检测要求的灵敏度,但是检测时间需要48小时。阈值法因为是采用DNA抗体的非特异序列免疫检测技术,不能特异性识别宿主残留DNA序列,且容易受到环境和操作人员的DNA污染,导致读值偏高。qPCR法具有序列特异性,灵敏度、准确度、精密度都好,还可以高通量筛选,但开发一个合格的q-PCR试剂检测宿主残留DNA并不是件容易的事情。有人会提出疑问,终产品中应该限定任何物种的DNA残余以确保安全性,所以阈值法是不是最合理的分析方法?
这里还需要强调一下宿主残余DNA检测的目的:
1.确认纯化工艺合理,能有效去除宿主DNA残余;
2.确认产品中杂质含量符合标准要求。非特异性的DNA检测结果不能区分究竟是生产中污染、检测污染、或是工艺缺陷引起的DNA残余,就无法为解决方案提供有效信息。在严格的生产体系中,残留DNA检测是解决工艺合理性问题,任何外源污染问题都归SOP或GMP管理体系解决。最后,经典的残留DNA检测方法灵敏度不同,qPCR法、DNA结合法、杂交法的检测限分别达到<1、<>
3、6pg/样品的水平(目前qPCR法灵敏度可达10fg),但是技术上限制,要求待测DNA片段分别不能小于50、150、600bp才能用于杂交法、q-PCR法、阈值法检测,而WHO和FDA可接受的DNA 限度内的片段长度<200bp。<>
由此可见,这三种方法中,qPCR法的适用性和技术指标最好。从qPCR技术原理来看,Taqman法要优于荧光染料随机掺入的SYBR Green法。正如前面介绍的USP修订内容,经过几年来对三种检测方法的系统研究和应用反馈,美国药典会将在新版药典中唯一推荐qPCR法作为生物制品残留DNA检测的标准方法。
系列4. 我国生物制品中残余DNA检测标准与技术
我国参照WHO、FDA和欧盟的标准,很早以前开始就对生物制品中残余DNA的含量进行限制。酵母、大肠杆菌表达的生物制品限定不超过10ng/剂,CHO和vero细胞表达的EPO、狂犬疫苗、乙肝疫苗等不超过100或10pg/剂。2010年版中国药典附录收录了DNA 探针杂交法和荧光染料法作为残余DNA检测方法。以地高辛标记斑点杂交法为代表的DNA杂交法因为操作复杂,耗时较长,且只能半定量,在2014年美国药典修订版征询意见稿中( PF40(2))中已经被剔除。以PicoGreen为代表的荧光染料法是基于荧光染料分子直接与双链DNA结合后,经带有荧光检测功能的酶标仪激发后产生荧光信号,因为不能检出单链DNA,且没有序列特异性,以及灵敏度较低(>300pg)等原因,早已被欧美药典摒弃。
我国2010版药典及2015版药典(附录IX B 外源性 DNA 残留量测定法)的征求意见稿中收载了的DNA探针杂交法和荧光染料法,而2009年美国USP就收录了杂交法、阈值法和qPCR法,到2015年底USP将只收录高灵敏度、高特异性的qPCR法作为残留DNA检测的推荐方法。由此可见qPCR法将成为国际公认的残留DNA检测方法,也可能会是我国下一版药典的主要修订内容。国内生物制品研发与生产企业,以及生产纯化填料的上下游企业,尽早建立以qPCR方法为基础的宿主残余DNA检测体系,将有利于改进工艺、提高产品质量,实现与欧美发达国家生物药品标准的接轨。
系列5. 国家标准物质库中的试剂盒技术性能
为了提升国内企业和监管部门对生物制品中宿主残余DNA的检测技术水平,实现与国际标准的接轨,中检院联合中科院、生产与研发企业开发了首个基于qPCR技术,具有自主知识产权的国产CHO宿主细胞DNA残留检测试剂盒。研发中除了常规方法学研究和稳定性考察,还开展了DNA碎片化、种属特异性、不同的仪器通用性、抗蛋白干扰、国外同类产品性能对比等研究,并在验证试验中考察了对国内多家生产企业和研发企业不同样本基质的适用性。由中检院采用国家标准物质CHO细胞DNA标定并验证的试剂盒提供了从样品提取、纯化到PCR检测的整套试剂,定量灵敏度达到10fg/rxn,DNA片段大于120bp,内标加样回收率在70~130%(新版美国USP的标准将调整为50~150%),可以为CHO细胞表达的不同品种、不同剂量的生物制剂提供灵活、准确的DNA限量检测。试剂盒具有2年保质期,并由联合研发单位中科院湖州营养中心提供免费技术咨询和操作培训服务,帮助企业尽快掌握该技术。中检院和中科院湖州营养中心的联合研发项目还将陆续推出E.coli、Yeast和vero细胞残余DNA检测试剂盒,为国内生物制药产业的发展、为监管部门制订国家标准提供技术支持。
来源:生物谷

为你推荐
 资讯
资讯 石药集团与阿斯利康成立合资公司,收到1000万美元里程碑付款
双方将在河北石家庄建设新一代生物制剂生产基地,该合资公司的初始业务将聚焦于为全球市场生产及供应双方约定的生物制剂药物原液(DS)。
2026-08-05 20:49
 资讯
资讯 全球首款!核心医疗人工心脏产品获欧盟CE认证
Corheart®6 可用于桥接心脏移植、短期心肌功能恢复治疗以及长期终点治疗,完整覆盖终末期心衰从短期生命支持到永久循环辅助的全场景临床需求
2026-08-05 20:37
 资讯
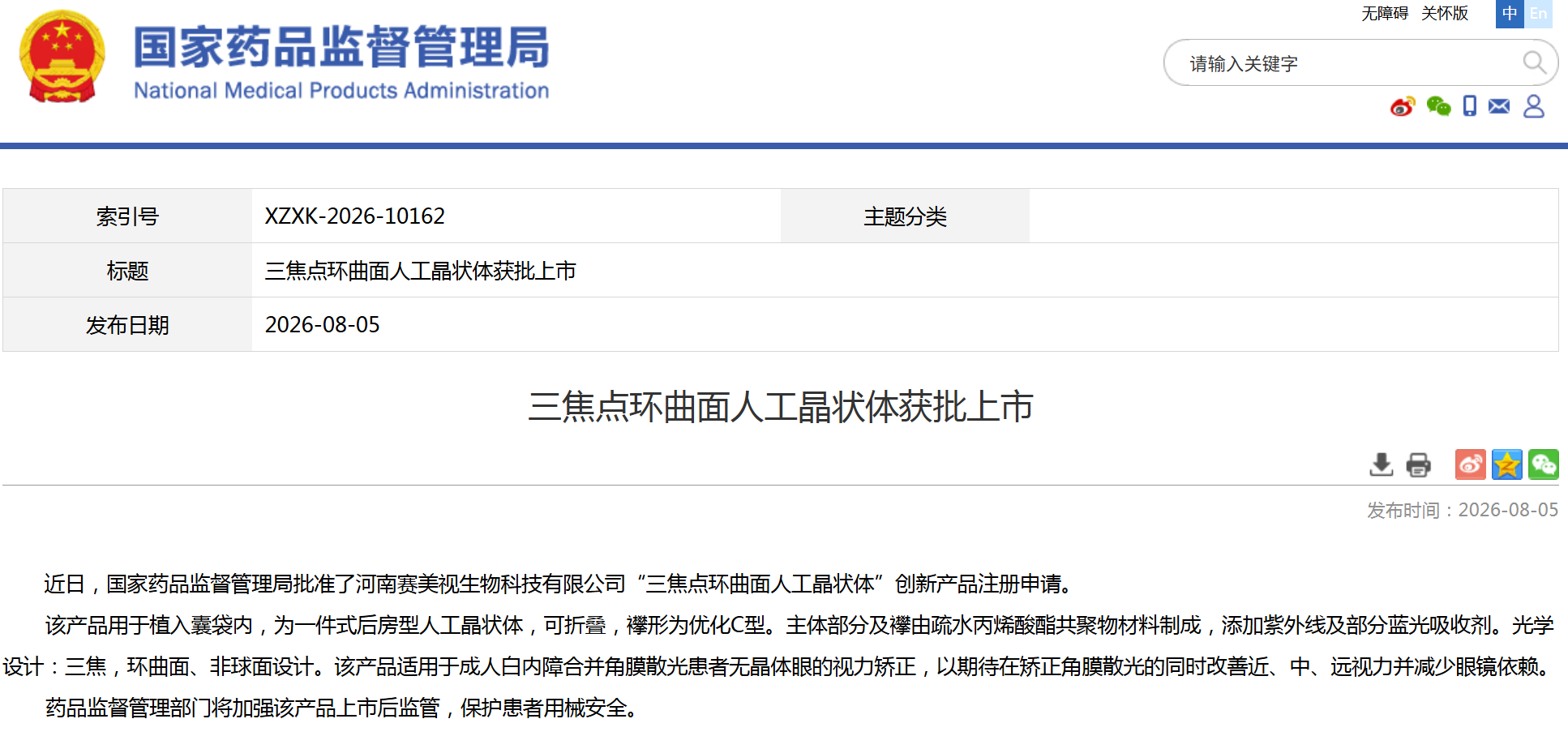
资讯 国家药监局批准3款创新医疗器械上市
近日,国家药品监督管理局批准了河南赛美视生物科技有限公司的“三焦点环曲面人工晶状体”和深圳北芯医疗科技有限公司的“心脏脉冲电场消融仪”、“一次性使用心脏脉冲电场消融...
2026-08-05 16:47
 资讯
资讯 80亿美元,全球两家核药公司合并
美东时间8月3日,Curium Pharma宣布与Lantheus Holdings, Inc 达成最终合并协议。Curium将以每股102 5美元现金收购Lantheus全部流通股,基础对价70亿美元,若2030年前达成...
2026-08-05 13:51
 资讯
资讯 章科:坚持为人民出政绩 推动医疗保障事业高质量发展
医疗保障制度依法保障每一位参保群众的医保权益,有力提高全社会医疗服务的可及性,有助于增进民生福祉、缩小贫富差距,促进社会公平和共同富裕。
2026-08-05 10:48
 资讯
资讯 康宁杰瑞与PathosAI达成22亿美元ADC授权协议,1.25亿美元首付款
8月4日,康宁杰瑞生物制药宣布其全资子公司江苏康宁杰瑞生物制药有限公司与美国临床阶段生物技术公司Pathos AI就TROP2 HER3双特异性抗体偶联药物JSKN016达成独家授权许可协议。
2026-08-04 22:14
 资讯
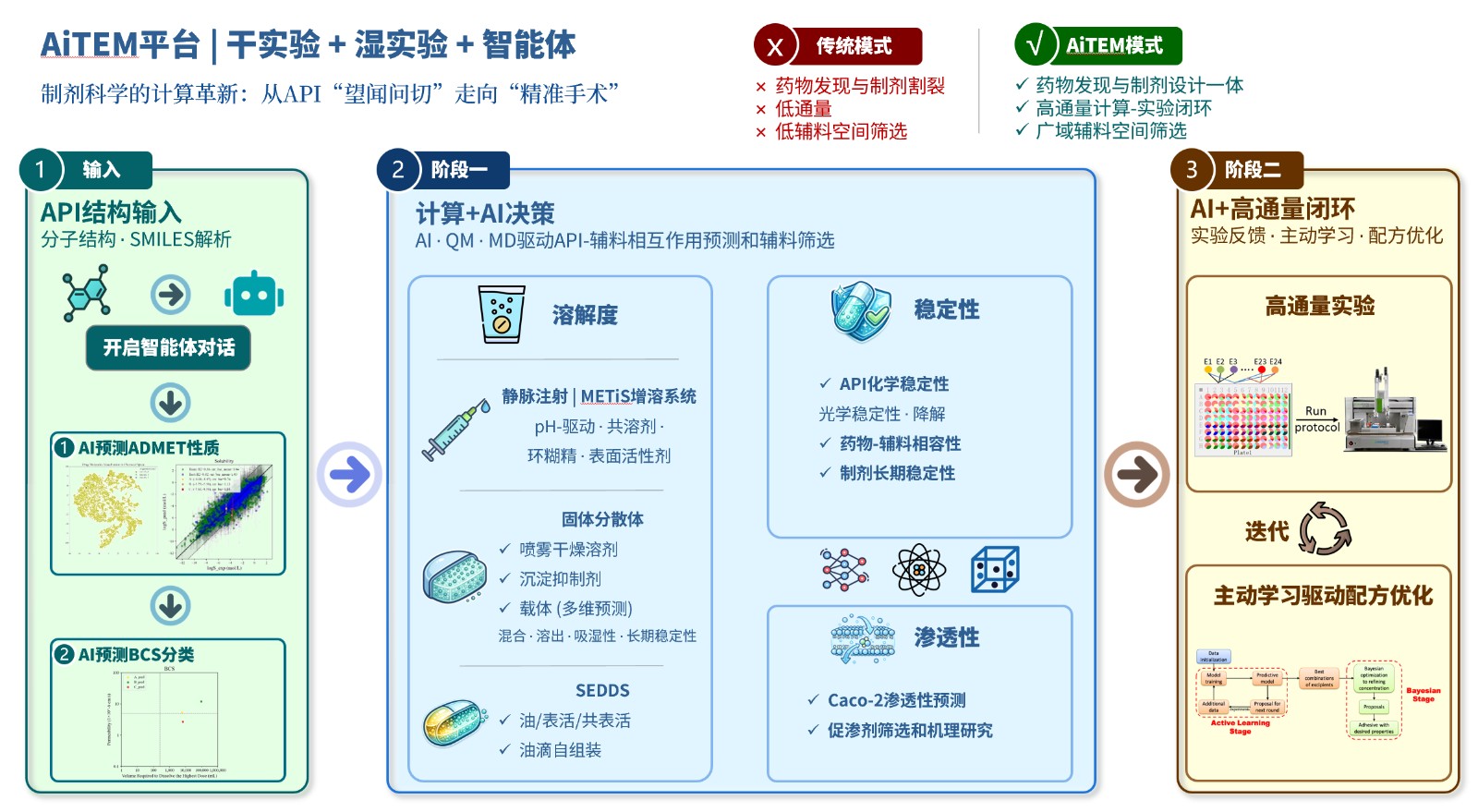
资讯 剂泰科技与恒瑞医药达成AI制剂合作
8月3日,剂泰科技发布自愿公告,宣布自主研发的AiTEM人工智能制剂创新平台将在恒瑞医药进行本地化部署,加速药物制剂配方开发效率。
2026-08-04 13:01

 资讯
资讯 东阳光药甘精胰岛素首批115.2万支发货美国
近日,东阳光药发布公告,首批甘精胰岛素注射液(商品名:Langlara)已于7月27日自湖北宜都生产基地分批启运美国,预计8月中下旬完成全部发货。首批计划发货115 2万支。
2026-08-04 11:37
 资讯
资讯 维利信LBL-024用于转移性结直肠癌获批IND
8月3日,维立志博发布公告,公司自主研发的PD-L1 4-1BB双特异性抗体维利信(LBL-024)联合用药治疗转移性结直肠癌(mCRC)的开放、多中心Ib II期临床试验申请已获得国家药品监...
2026-08-04 10:22
 资讯
资讯 药明康德半年业绩超预期,大幅上调全年业绩指引
预计2026年公司整体收入由人民币513-530亿元上调至人民币585-605亿元,上调幅度约14%,持续经营业务收入同比增速由18%-22%上调至35%-39%。药明康德计划向全体股东每10股派发现金红利5 1元(含税)。
2026-08-03 20:46



 资讯
资讯 国家医保局召开全国深化医保基金管理突出问题专项整治工作中期推进会
7月31日,国家医保局会同最高人民法院、最高人民检察院、公安部、财政部、国家卫生健康委、税务总局、市场监管总局、国家中医药局、国家药监局,10部门联合召开全国深化医保基金...
2026-08-03 10:40
 资讯
资讯 第12批国采,哪家企业中选品种最多?
本次集采65个品种全部采购成功,全国共4 5万家医药机构报量,327家企业的521个产品获得拟中选资格,覆盖抗感染、抗肿瘤、抗血栓、降血糖、降血压、降血脂、风湿免疫、消炎镇痛...
2026-08-02 19:58