
今日(11月8日),国家药监局药品审评中心发布关于再次公开征求《临床急需药品附条件批准上市技术指导原则》意见的通知,以鼓励以临床需求为核心的药物创新,加快具有临床价值的临床急需药品上市。

根据国家药监局的要求,我中心组织起草了《临床急需药品附条件批准上市技术指导原则》。该指导原则曾以技术指南的形式于2017年12月20日在原食药总局网站征求意见,后续在卫建委等相关部委、国家局相关司局及直属单位范围内征求意见。现根据《中华人民共和国药品管理法》、《中华人民共和国疫苗管理法》的最新要求以及《药品注册管理办法(征求意见稿)》的相关内容,结合征求到的各方意见,进行了修改。现再次向社会公开征求意见。
欢迎社会各界对征求意见提出宝贵意见和建议,并及时反馈给我们。征求意见请于2019年11月13日17:00前反馈。
您的反馈意见请发到以下联系人的邮箱:
联系人:王洪航、赵晨阳
联系方式:wanghh@cde.org.cn、zhaochy@cde.org.cn
国家药品监督管理局药品审评中心
2019年11月08日
附件一:临床急需药品附条件批准上市技术指导原则(征求意见稿)
一、前言
为落实《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),鼓励以临床需求为核心的药物创新,加快具有临床价值的临床急需药品上市,根据《中华人民共和国药品管理法》、《中华人民共和国疫苗管理法》、《药品注册管理办法(征求意见稿)》,借鉴国际经验,结合我国药品审评工作实践,制定本指导原则。
附条件批准上市是指用于治疗严重危及生命且尚无有效治疗手段的疾病以及罕见病的药品、公共卫生方面急需的药品,现有临床研究资料尚未满足常规上市注册的全部要求,但药物临床试验已有数据显示疗效并能预测其临床价值,因临床急需,在规定申请人必须履行特定条件的情况下基于替代终点、中间临床终点或早期临床试验数据而批准上市。其目的是缩短药物临床试验的研发时间,提早应用于无法继续等待的急需病人。附条件批准上市不包括因临床试验设计或执行过程中存在缺陷而不能达到上市许可要求的情况。
通常,附条件批准上市药品的药学、药理毒理学要求与常规批准上市药品相同;对于公共卫生方面急需的药品或应对重大突发公共卫生事件的药品,可根据具体情况,结合药品的获益-风险进行评价。
本指导原则适用于未在中国境内上市销售,但临床急需的中药、化学药和生物制品。
申请人在获得附条件批准上市后,需按照所附的特定条件,开展新的或继续正在进行的临床试验,这些临床试验通常是以确认预期的临床获益为目的的确证性临床试验,为常规上市提供提供充足证据。
二、附条件批准上市的基本条件
(一)符合以下情形的临床急需药物,可申请附条件批准:
1. 目标适应症为严重且危及生命的疾病,其现有治疗手段具有未满足的临床需求,药物临床试验已有数据显示其疗效并能预测其临床获益的;
2. 治疗罕见病的临床急需药品,境外已批准上市或境内已有临床数据显示其疗效并能预测其临床获益的;
3. 公共卫生方面急需的药品,药物临床试验已有数据显示其疗效并能预测其临床获益的;
4. 应对重大突发公共卫生事件急需的疫苗或国家卫生行政部门认定急需并被依法列入特别审批程序的其他疫苗,经评估获益大于风险的。
(二)未被满足的临床需求通常包括以下情况:
1. 无批准可用的治疗方法。
2. 有可用的治疗方法:当一种疾病存在可用的治疗方法,如果新的治疗药物能满足下述条件之一,通常也被认为可解决未满足的临床需求:
(1)与现有疗法相比,对疾病的预后有明显改善作用;
(2)用于对现有疗法不耐受或无疗效的患者可取得明显疗效;
(3)可以与现有疗法不能联用的其他关键药物或治疗方式有效地联用并取得明显疗效;
(4)疗效与现有疗法相当,但可避免现有疗法的严重毒性,或降低有害的药物相互作用,或显着改善病人的依从性;
3. 解决新出现或预期会发生的公共卫生需求。
三、附条件批准药物的疗效评价
对于临床急需药物,可基于替代终点、中间临床终点或早期临床试验数据而附条件批准上市。申请人应说明所选择的替代终点、中间临床终点或选择早期临床试验数据与预期的临床获益之间的相关性、合理性,并提供相应的证据。
1. 有效性评价终点指标
通常用于药物有效性评价的指标应为临床终点。临床终点是指可以直接测量药物疗效的特征或变量,即药物对患者感觉(例如症状缓解)、功能(例如运动性改善、延缓或阻止功能衰退等)或生存影响的直接评价。
用于附条件批准临床急需药品上市的有效性评价终点通常有两类:
(1)很可能预测临床获益的替代终点。替代终点一般是一个生物标志物,可以是实验室检查项目、放射影像学、体征或其他指标,其本身并不衡量临床获益,但可以预测临床获益。依据其对临床获益的预测能力,替代终点可以是已知可以合理预测临床获益的指标,或者是很可能预测临床获益的指标。
(2)可以早期评估临床获益的中间临床终点。中间临床终点一般是指在治疗慢性疾病的临床获益评价中,通常认为短期临床获益很可能预测长期临床获益。例如,治疗多发性硬化病的药物在获得常规批准时需要提供2年的用药临床疗效评价,而在附条件批准时,中间临床终点指标则是1年的用药疗效评价。
在确证性临床试验中,应用替代终点指标或中间临床终点指标的研究结果可以合理预测该产品很可能具有疗效和临床获益的,可给予附条件批准上市。
2.早期临床试验数据
早期临床试验数据通常是指在开展确证性临床试验前所获得的临床数据。根据早期临床试验数据,可合理预测或判断其临床获益的,可以在完成确证性临床试验前给予附条件批准上市。
3. 境外临床试验数据
境外已批准上市的治疗罕见病的临床急需药品,基于境外临床试验数据,可以在完成种族敏感性试验前给予附条件批准上市。
四、附条件批准上市的申请
(一)沟通交流
1. 临床试验开展前
申请人在附条件批准上市的临床试验开展前,应与国家药品监督管理局药品审评中心(以下简称药审中心)进行沟通交流,以明确下列问题:
(1)临床试验中所选择的替代终点指标的合理性及其可合理预测临床获益的标准或
(2)临床试验中中间临床终点指标的选择及其可合理预测临床获益的标准或
(3)对早期临床试验数据的要求,包括疗效评价的指标和标准,可合理预测临床获益的依据。
(4)上市后临床试验的设计和实施计划。
(5)其他附条件批准的前提条件,包括药学、药理毒理学研究等。
沟通会议纪要将作为附条件批准上市申请的受理、立卷审查和审评审批的重要依据。
2. 提交上市申请前
申请附条件批准上市前,申请人应当就已有临床试验数据、药学和药理毒理学数据、申请附条件批准上市的意向以及上市后继续完成的研究工作、上市后风险控制计划等与药审中心进行沟通交流。经沟通交流确认后,可提出药品上市注册申请。
(二)提交上市申请的要求
根据替代终点指标而附条件批准上市的,申请人应在申请上市许可时提交承诺的上市后确证性临床研究计划和方案以及完成期限、上市后风险控制计划等。
根据中间临床终点而附条件批准上市的,申请人需承诺按已确定的临床试验方案完成临床试验并提交上市后风险控制计划。
根据早期临床试验数据附条件批准上市的,申请人应在申请上市许可时提交后续临床研究计划和方案以及完成期限、上市后风险控制计划等。
已在境外批准上市的治疗罕见病的药品,申请人需按照我国相关要求提交支持其境外批准上市的临床试验数据以及种族敏感性研究的计划和方案、风险控制计划。
在上市申请审评期间,申请人与药审中心可进一步就上述内容讨论确定并达成一致意见,申请人应按时完成所有承诺的临床试验。
五、附条件批准上市的上市后要求
对附条件批准的药品,国家药监局将以《药品注册批件》附件的形式将药品上市所附条件以及申请人承诺的研究和完成期限通知药品上市许可持有人。
在药品说明书【适应症】和【临床试验】项下,应注明本品为基于替代终点(或中间临床终点或早期临床试验数据)获得附条件批准上市,尚未获得临床终点数据,有效性和安全性有待上市后进一步确证;【批准文号】项下应注明“附条件批准上市”字样。药品标签中相关内容应与说明书保持一致。
药品上市许可持有人应当在药品上市后采取相应的风险管理措施,保证患者用药安全。并在规定期限内按要求完成承诺的药物临床试验等相关研究,以补充申请方式报药审中心申请常规批准上市。
附条件批准上市后开展新的或继续进行的临床试验,仍需符合《药物临床试验质量管理规范》的相关要求。
六、附条件批准上市的撤销
有以下情形之一的,国家药品监督管理局依法处理,直至注销附条件批准药品的药品注册证书:
(一)逾期未按要求完成后续相关研究且无合理理由的;
(二)要求证实药品预测临床获益的试验未能证实该获益的;
(三)上市后临床研究结果经审评不能证明获益大于风险的;
(四)其他不符合继续上市条件的情形。
附件二:《临床急需药品附条件批准上市技术指导原则》起草说明
一、背景
为落实《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),鼓励以临床需求为核心的药物创新,加快具有临床价值的临床急需药品上市,根据《中华人民共和国药品管理法》、《中华人民共和国疫苗管理法》、《药品注册管理办法(征求意见稿)》,借鉴国际经验,结合我国药品审评工作实践,制定本指导原则。
本指导原则共分为6个部分,分别为前言、附条件批准上市的基本条件、附条件批准药物的疗效评价、附条件批准上市的申请、附条件批准上市的上市后要求、附条件批准上市的撤销。
本指导原则(原技术指南)于2017年12月20日-2018 年1月15日期间在原国家食品药品监督管理总局网站面向全社会征求意见,根据反馈意见进行了修订。后续在卫建委等相关部委、国家局相关司局及直属单位范围内征求意见并进行修订。现根据《中华人民共和国药品管理法》、《中国人民共和国疫苗管理法》《药品注册管理办法(征求意见稿)》再次进行修订。需要指出的是,本指导原则是根据国内临床试验研究现状和现有药品审评工作实践制定,随着临床研究经验积累和审评工作的改革,本指导原则亦可进行相应的调整和完善。
二、征求意见反馈信息情况
(一)网上征求意见反馈
2017年12月20日至2018 年1月15日征求意见期间,共收到19份有效反馈,其中个人2份,企业15份,其他组织和团体2份。整理出反馈意见96条,其中总体意见8条,第一章总则相关意见22条,第二章基本条件相关意见16条,第三章疗效指标选择相关意见21条,第四章责任和义务相关意见29条。
反馈意见包括以下内容:指南适用范围、罕见病的定义或目录、企业申请有条件批准上市的时间、早期或中期临床试验数据的范围、境外上市境内未上市的治疗罕见病药品的有条件批准、有条件批准上市后撤市的规定等问题。
(二)定向征求意见反馈
2018年定向征求意见期间,收集到卫建委反馈意见3条,包括以下内容:临床急需药品的遴选、上市后研究的要求及再评价、上市后研究及临床应用的风险控制等。
(三)药审中心内部征求意见反馈
2019年10月28日、11月4日分别以邮件和会议的形式在药审中心内部征求意见,收集到的反馈意见包括以下内容:附条件批准药品的药学和药理毒理学的要求、附条件批准上市药品的说明书和标签描述等。
三、修订说明
主要修订内容包括:
(一)标题由“临床急需药品有条件批准上市的技术指南”修订为“临床急需药品附条件批准上市技术指导原则”。
(二)指南适用范围统一为“未在中国境内上市销售,但临床急需的中药、化学药和生物制品。”
(三)增加了附条件批准上市药品的药学、药理毒理学要求。
(四)附条件批准上市的基本条件增加了疫苗的相关描述。
(五)附条件批准上市的申请部分,细化了“沟通交流”和“提交上市申请的要求”部分的内容。
(六)增加了附条件批准上市药品的《药品注册批件》、说明书和标签的描述规定,明确了以补充申请的形式提交附条件批准上市后完成的临床试验以推动药品常规批准上市。
来源:CDE

为你推荐
 资讯
资讯 云顶新耀年销超14亿的当家花旦迎来新的挑战者
5月21日晚间,石药集团发布公告宣布,其开发的布地奈德肠溶胶囊(4mg)已获得国家药品监督管理局颁发的药品注册批件,用于治疗具有疾病进展风险的原发性免疫球蛋白A肾病成人患者...
2026-05-24 22:38
 资讯
资讯 5月20日起,提交注册申请的品种按照《M13A:口服固体速释制剂的生物等效性》执行
自本公告发布之日起,提交注册申请的品种均适用M13A指导原则(含问答文件)。对于已按照既往临床试验方案和统计分析计划开展生物等效性试验,并在本公告发布之日起12个月内申报...
2026-05-23 11:56
 资讯
资讯 呋喹替尼联合疗法获批晚期肾细胞癌患者二线治疗新适应症
5月21日,和黄医药宣布,其自主研发创新药物呋喹替尼获得中国国家药品监督管理局(NMPA)批准,联合信迪利单抗用于治疗既往接受血管内皮生长因子受体酪氨酸激酶抑制剂(VEGFR-...
2026-05-22 15:56
 资讯
资讯 刚果(金)与乌干达埃博拉疫情:国际SOS发布企业员工支持建议
近日,世界卫生组织(WHO)宣布,当前影响刚果民主共和国和乌干达的埃博拉疫情构成“国际关注的突发公共卫生事件”(PHEIC)。
2026-05-22 13:47
 资讯
资讯 早筛 戒烟 筑牢膀胱癌第一道防线
每年的5月是“膀胱关爱月”,根据国家癌症中心发布的《2024年全国癌症报告》显示,膀胱癌位居我国恶性肿瘤发病第 11 位。2022年中国膀胱癌发病率为7 3 10万人,死亡率为4...
文/刘思 2026-05-22 13:31
 资讯
资讯 再鼎医药总裁兼首席运营官离职
5月22日早间,再鼎医药发布公告称,公司董事会已于5月18日决定,Josh Smiley不再担任公司总裁兼首席运营官,相关决定自当日起生效,其在公司的最后任职日期为5月22日。
2026-05-22 11:45

 资讯
资讯 共建多元支付,加速创新可及:镁信健康与辉瑞中国达成战略合作
双方将依托长期合作基础,以多元支付为基石,以数据洞察和AI技术为引擎,加速前沿创新药落地中国,全方位提升患者用药可及性与健康服务体验,助力构建多层次医疗保障体系。
2026-05-21 17:57
 资讯
资讯 拜耳可申达(非奈利酮片)在中国获批用于LVEF≥40%的心力衰竭成人患者
拜耳宣布中国国家药品监督管理局(NMPA)批准高选择性非甾体类盐皮质激素受体拮抗剂可申达(非奈利酮片)用于射血分数(LVEF)≥40%的心力衰竭成人患者,以降低心血管死亡、因心...
2026-05-21 17:51
 资讯
资讯 冯慧宇教授:从“活下去”到“活得好”,重症肌无力迈入精准治疗时代
作为深耕重症肌无力领域数十年的临床专家,她用一个个真实的患者故事,揭示了这个有着 300 多年历史的古老疾病在当代诊疗中的核心痛点,并分享了对创新药发展和患者全程管理的...
文/张蓉蓉 2026-05-21 15:48
 资讯
资讯 2026年“家庭健康促进计划-健康同行1+1公益项目” 在西安医学院正式启动
5月18日,由中国妇女发展基金会主办,陕西省妇联、陕西妇女儿童发展基金会、西安医学院协办,默沙东公益支持的“家庭健康促进计划-健康同行1+1公益项目西安高校健康跑暨2026启动...
2026-05-21 15:37
 资讯
资讯 爱尔眼科补交税款及滞纳金5.24亿元
5月20日,爱尔眼科(300015 SZ)发布公告称,其根据国家税收法律法规相关要求进行自查后,需补缴税款3 48亿元,并支付滞纳金1 76亿元,合计金额达5 24亿元。目前,上述税款...
2026-05-21 15:36
 资讯
资讯 复宏汉霖引进第三代口服EGFR-TKI产品
近日,复宏汉霖宣布与江苏正大丰海制药有限公司及其旗下江苏创特医药科技有限公司达成战略合作。根据约定,复宏汉霖将获得由江苏创特自主研发的口服小分子三代表皮生长因子受体...
2026-05-21 11:28
 资讯
资讯 冠脉支架国家集采第二轮接续采购在天津开标,27个产品拟中选
5月20日,国家组织冠脉支架集中带量采购第二轮接续采购在天津开标。共15家企业的30个产品参与投标,投标企业全部中选,27个产品获拟中选资格。
2026-05-20 21:05

 资讯
资讯 因美纳发布年度企业责任报告,持续提升基因组学可及性,加速拓展全球影响力
报告重点介绍了公司在推动基因组学公平可及方面取得的持续进展,以及为全球患者、社区和医疗系统带来可衡量的影响
2026-05-20 18:14
 资讯
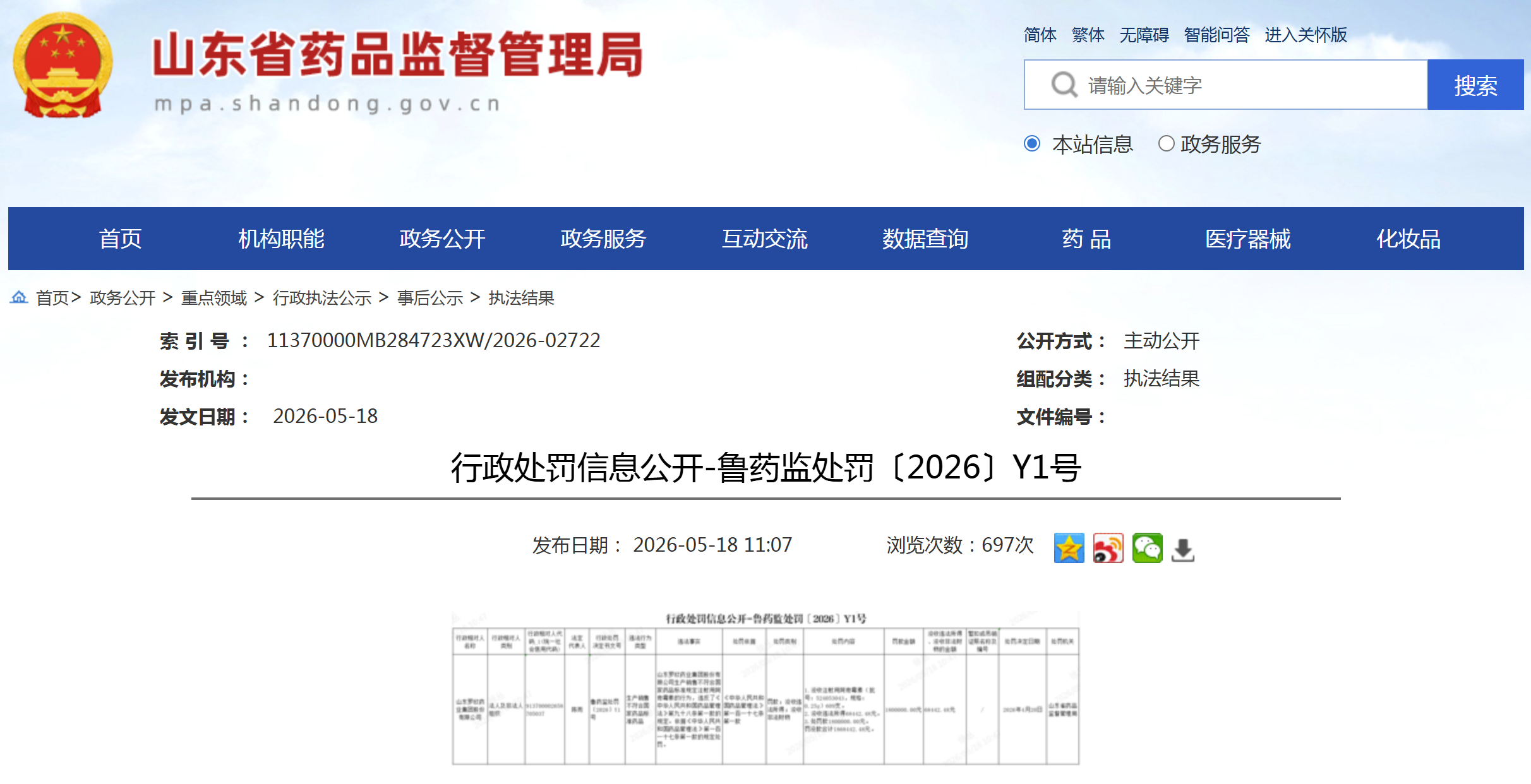
资讯 罗欣药业因生产劣药被处罚186万元
5月18日,山东省药监局发布的行政处罚信息显示,罗欣药业因生产销售不符合国家药品标准的注射用阿奇霉素,被处以没收药品609支、没收违法所得6 8万元并处罚款180万元,罚没款合...
2026-05-20 16:01
 资讯
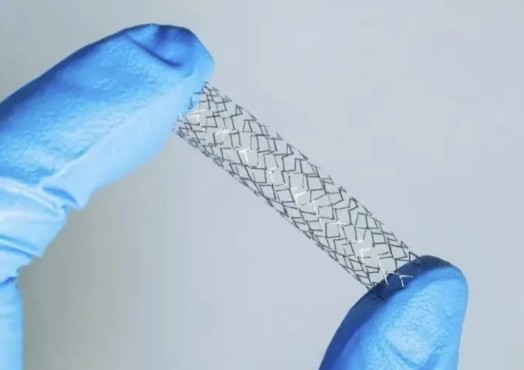
资讯 集采心脏支架临床使用超千万
2020年,心脏支架成为首批国家组织高值医用耗材集中带量采购品种,国内外企业的10个临床主流产品中选,普遍降至1000元以下,我国心脏支架基本告别“万元时代”。
2026-05-20 13:46
 资讯
资讯 我国启动首个128通道全植入式脑机接口系统多中心临床试验
5月18日,我国正式启动首个128通道全植入式脑机接口系统多中心临床试验,这项试验由首都医科大学附属北京天坛医院担任组长单位。
2026-05-19 21:08
 资讯
资讯 国产口服小分子GLP-1减重适应症完成III临床试验,即将申报NDA
5月18日,成都闻泰医药科技股份有限公司宣布,公司自主研发的每日一次口服小分子GLP-1受体激动剂VCT220片,在中国超重或肥胖受试者中的关键Ⅲ期临床试验取得积极顶线结果,计划...
2026-05-19 18:07