
为规范我国生物类似药的科学研发与评价,推动生物医药行业的健康发展,满足生物治疗产品的可获得性和可支付性,10月29日,药品审评中心(CDE)网站最新发布《生物类似药研发与评价技术指导原则(征求意见稿) 》(以下简称“《意见稿》”)。
此意见稿传递出了很多利好信息,有业界人士表示,这标识着业界望穿秋水的生物类似药(另有一说“生物仿制药”)政策大门即将开启,中国生物制药行业有望迎来一个春天。
也有业内人士分析,该指导原则的标准与国际接轨,未来获批的生物类似物必须符合国际标准,这对企业提出了更高的要求,这也要考验企业是否能够承受大量时间和资金的投入。
近年来,生物技术药占全球医药市场的比重正在逐步提高,从2006年的14%上升到2014年的23%;投入市场的生物技术类药不断增多,1989年仅13个,2012年已达210个;全球畅销的100强药物中,2006年生物药占比21%,2020年将达到52%,增长迅猛。“全球医药市场正在上演由小分子向生物药的戏剧化转变。”国家食品药品监督管理总局南方医药经济研究所副所长陶剑虹指出。
随着一批生物药品专利在未来5年内密集到期,生物类似物市场将迎来历史机遇。据evaluatePharm预测,全球生物类似物市场将从2010年的2.23亿美元,增长到2015年的100亿美元,2020年更是将超过200亿美元,10年间有望达到90倍的增长,复合增长率达到56%。然而,由于我国尚无针对生物类似物的相关法规和技术原则,国内企业从事生物类似物开发时面临诸多不确定因素,申报时需按照创新药的流程进行,但临床试验也按照类似药的要求去操作,导致最后试验结果不符合审评要求,大大浪费了资源,造成巨大损失。
齐鲁制药药物研究院首席科学家杨建国博士也表示,近几年,国内涉足生物类似物的企业众多,不少品种出现申报企业扎堆的情况。然而,开发生物类似物对企业的要求远高于化学药。开发生物类似物需要具备诸多基本能力,如细胞株和工艺开发、临床前分析鉴定、工艺放大和生产、临床研究、与药监部门的沟通等等,获批上市后还要面临销售与市场、支付问题和上市后临床研究等方面的挑战,而此次生物类似物研究技术指导原则的制定出台无疑将为摸着石头过河的相关企业指引方向。
附:《生物类似药研发与评价技术指导原则(征求意见稿)》
1.前言
生物药在许多威胁生命的疾病治疗方面已显示出明显的临床优势。随着原研生物药专利到期及生物技术的不断发展,生物类似药的研发越来越受到重视,有助于提高医药产品的可获得性及可及性。为规范生物类似药的研发与评价,推动生物医药行业的健康发展,制定本指导原则。 生物类似药的研发与评价应当遵循本指导原则,并应符合国家药品管理相关规定的要求。
2.定义及适用范围
本指导原则所述生物类似药是指,在质量、安全性和有效性方面与已获准注册的原研药具有相似性的治疗用生物制品。 生物类似药候选药物的氨基酸序列应与原研药相同。对研发过程中采用不同于原研药所用的宿主细胞、表达体系等的,需进行充分验证。
本指导原则适用于结构和功能明确的治疗用重组蛋白质制品。对聚乙二醇等修饰的产品及抗体偶联药物类产品等,按生物类似药研发时应慎重考虑。
3. 参照药
3.1定义
本指导原则所述参照药是指,已获得国家药品监督管理当局生物类似药研发与评价技术指导原则批准注册的,在生物类似药研发过程中与之进行比对研究用的原研产品,包括生产用的或由成品中提取的活性成分。
3.2参照药的选择
比对试验研究用的参照药应当是在我国已经注册的产品,并证明安全有效。 研发过程中各阶段所使用的参照药,应尽可能使用相同批号来源的产品。对不能通过商业途径在国内获得的,可以考虑其他合适的途径;对临床比对试验研究用的,还应符合国家的其他相关规定。 比对试验研究必须用活性成分的,可以采用适宜方法分离,但需验证这些方法对活性成分的结构和功能等质量特性的影响。 按生物类似药批准的产品原则上不可用作参照药。
4.研发和评价的基本原则
4.1比对原则
生物类似药研发是以比对试验研究证明其与参照药的相似性为基础,支持其安全、有效和质量可控。 每一阶段的每一个比对试验研究,均应与参照药同时进行,并设立相似性的评价方法和标准。
4.2逐步递进原则
研发应采用逐步递进的顺序,分阶段证明候选药与参照药的相似性。比对试验结果无差异或者差异很小,评判为相似的,可以减免后续的部分比对试验研究;对存在较大差异或不确定因素的,需评估对产品的影响,在后续研究阶段还必须选择敏感的技术和方法设计有针对性的比对试验进行研究,并评价对产品的影响。
4.3一致性原则
比对试验研究所使用的样品应当保持前后的一致性。对候选药,应当为生产工艺确定后生产的产品,或者其活性成分。对不同批或者工艺、规模和产地等发生改变的,应当评估对产品质量的影响,必要时还需重新进行比对试验研究。 比对试验研究的方法和技术应尽可能与参照药所使用的保持一致,至少在原理上应一致。在研发过程中,对存在的差异,应当选择敏感的技术和方法设计针对性的比对试验研究,并进行验证评估其适用性和可靠性。
4.4相似性评价原则
对全面的药学比对试验研究显示候选药与参照药相似,并在非临床阶段进一步证明其相似的,后续的临床试验可以考虑仅开展临床药理学比对试验研究;对不能证明相似性的,后续还应开展针对性的研究或临床安全有效性的比对试验研究。药学比对试验研究显示的差异对产品有影响并在非临床比对试验研究结果也证明的,对继续研发的,后续应开展系统的临床比对试验。对临床比对试验研究结果判定为相似的,可按本指导原则进行评价。
5.药学研究和评价
5.1一般考虑
比对试验研究应使用足够批次进行,一般情况下应进行至少各三批的比对试验研究。研究中,应尽可能使用经过验证的、灵敏的、先进的分析技术和方法检测候选药与参照药之间可能存在的差异。
5.2工艺研究
候选药的生产工艺需根据产品特点设计,应尽可能与参照药一致,尤其是工艺步骤的原理和先后顺序及中间过程控制的要求,如纯化、灭活工艺等。
5.3分析方法
比对试验研究的分析方法应尽可能与参照药所用的一致。对采用先进的、更为敏感的技术和方法,其基本原理应当相似,并应进行充分的验证。对某些关键的质量属性,应采用多种方法进行比对试验研究。
5.4特性分析
根据参照药的信息,分析、建立每一个质量特性与临床效果的相关性,并设立判定相似性的限度范围。对特性分析的比对试验研究结果综合评判时,应根据各质量特性与临床效果相关的程度确定评判相似性的权重,并设定标准。
5.4.1理化特性
理化鉴定应包括采用适宜分析方法确定一级结构和高级结构(二级/三级/四级)以及其它理化特性。还应考虑翻译后的修饰可能存在的差异,如氨基酸序列N端和C末端的异质性、糖基化修饰(包括糖链的结构和糖型等)的异同。应采用适当的方法对修饰的异同进行比对试验研究,包括定性和定量分析。
5.4.2生物学活性
生物学活性的比对试验研究对评判候选药与参照药有无显着功能差异具有重要意义。比对试验研究采用的技术和方法应尽可能与参照药所用的一致,至少在原理上应当相同。对具有多重生物活性的,还应当进行相关活性的比对试验研究,并分别设定相似性的评判标准;对相似性的评判,应根据各种活性与临床效果相关的程度确定评判相似性的权重,并设定标准。
5.4.3纯度和杂质
应尽可能采用参照药所用的技术和方法进行比对试验研究。对纯度的测定,应从产品的疏水性、电荷和分子大小变异体及包括糖基化在内的各类翻译后修饰等方面考虑适宜的技术和方法进行研究;对杂质的比对试验研究,应从工艺的差异、宿主细胞的不同等方面考虑适宜的方法进行。 对杂质图谱的差异,尤其是出现了新的成分,应当进行分析并确证,制定相应的质量标准,必要时在后续的比对试验研究中,还应设计有针对性的技术和方法研究对有效性、安全性包括免疫原性的影响。
5.4.4免疫学特性
对具有免疫学特性的产品的比对试验研究应尽可能采用与参照药相似原理的技术和方法。对具有多重免疫学特性的,还应当进行相关活性的比对试验研究,并分别设定相似性的评判标准;对相似性的评判,应根据各种特性与临床效果相关的程度确定评判相似性的权重,并设定标准。 对抗体类的产品,应对其Fab、Fc段的功能进行比对试验研究,包括定性、定量分析其与抗原和FcRn、Fcγ、c1q等各受体的亲和力、CDC活性、ADCC活性等。应根据产品特点选择适当的项目列入质量标准。 对调节免疫类的产品,应对其同靶标的亲和力、引起免疫应答反应的能力进行定性或者定量比对试验研究。应根据产品特点选择适当的项目列入质量标准。
5.5质量指标
候选药质量指标的设定及标准应尽可能与参照药设定的一致,并应符合药品管理相应法规的要求。对需增加指标的标准,应根据多批次产品的检定数据,用统计学方法分析确定,并结合稳定性数据等分析评价其合理性。
5.6稳定性研究
按照有关的指导原则开展稳定性的研究。对比对试验研究,应尽可能使用与参照药有效期相近的候选药进行。对加速试验或强制降解稳定性试验,应选择敏感的条件同时处理后进行比对试验研究。
5.7其它研究
5.7.1细胞基质
应考虑参照药所使用的细胞基质,也可采用当前常用的细胞基质。对与参照药不一致的,需进行充分的验证,并证明与有效性、安全性等方面无临床意义的差别。
5.7.2制剂处方
应尽可能与参照药一致。对不一致的,应有充足的理由并应进行处方筛选研究。
5.7.3规格
原则上应与参照药一致。对不一致的,应有恰当的理由。
5.7.4内包装材料
应当使用与参照药同类材质的内包装材料。对不同的,应有相应的研究结果支持。
5.8药学研究相似性的评价
对药学研究结果相似性的评判,应根据与临床效果相关的程度确定评判相似性的权重,并设定标准。
(1)对综合评判候选药与参照药之间的差异很小或无差异的,可判为相似;(2)对研究显示候选药与参照药之间存在差别,且无法确定对药品安全性和有效性影响的,应设计针对性的比对试验研究,以证实其对药品安全性和有效性是否有影响;(3)对研究显示有差异,评判为不相似的,不宜继续按生物类似药研发。 不同种类的重组蛋白,甚至是同一类蛋白,由于其疗效机制不同,质量属性差异的权重也是不一样的,分析药学质量相似性时要予以考虑。
6.非临床研究和评价
6.1一般考虑
应进行非临床比对试验研究,尤其是对所采用的细胞基质及杂质等不同于参照药的。对药学比对试验研究显示候选药和参照药无差别或仅有微小差别时,可仅开展药效动力学(简称药效,PD)、药代动力学(简称药代,PK)和免疫原性的比对试验研究。
6.2药效学
应开展系统的体外及体内药效学比对试验研究。对具有多重生物活性的,还应当进行相关活性的比对试验研究,并分别设定相似性的评判标准;对相似性的评判,应根据各种活性与临床效果相关的程度确定评判相似性的权重,并设定标准。 体内药效学比对试验研究应尽可能选择参照药采用的相关动物种属或模型进行。 比对试验研究方法和检测指标,应尽可能与参照药一致;应选择多批次有代表性的产品进行,以分析批间差异的影响。
6.3药代动力学
应选择相关动物种属开展多个剂量组的单次给药和重复给药的药代动力学研究。单次给药药代动力学试验应单独开展;重复给药的药代动力学试验可结合在PK/PD研究中或者重复给药毒性试验中进行。对受试药的药代动力学试验方法影响药物效应或者毒性反应评价的,应进行独立的重复给药比对试验研究来评估PK特征变化。
6.4免疫原性
采用的技术和方法应尽可能与参照药所使用的一致,对采用其它相似方法的,还应进行验证。抗体的检测包括筛选、确证、定量和定性,并研究与剂量和时间的相关性。必要时应对所产生的抗体进行交叉反应测定,对有差异的还应当分析其产生的原因。对量化的比对试验研究结果,应评价其对药代动力学的影响。 对所采用的细胞基质、修饰及杂质等不同于参照药的,还应设计针对性的比对试验研究。 在免疫原性试验中可同时观察一般毒性反应。
6.5重复给药毒性试验
对仅开展药效、药代动力学及免疫原性比对试验研究,其研究结果显示有差异且可能与安全性相关的,还应进行毒性比对试验研究。
对毒性比对试验,应进行至少一项相关动物种属的至少4周的研究,持续时间应足够长以能监测到毒性和/或免疫反应。研究指标应关注与临床药效有关的药效学作用或活性,并应开展毒代动力学研究。对有特殊安全性担忧的,可在同一重复给药毒性研究中纳入相应观察指标或试验内容,如局部耐受性等。 比对试验研究用的动物种属和模型、给药途径及剂量应尽可能与参照药一致。对选择其他的,应当进行论证。对参照药有多种给药途径的,必要时应逐一开展研究;对剂量的选择,应尽可能选择参照药暴露毒性的剂量水平,候选药剂量还应包括生物活性效应剂量和/或更高剂量水平。
6.6其它毒性试验
对药学及非临床比对试验研究显示有差异且不确定其影响的,应当开展有针对性的其它毒性试验研究,必要时应进行相关的比对试验研究。
6.7 非临床研究相似性的评价
对非临床研究结果相似性的评判,应根据与临床效果相关的程度确定评判相似性的权重,并设定标准。
(1)对综合评判候选药与参照药之间的差异很小或无差异的,可判为相似;(2)对研究显示候选药与参照药之间存在差别,且无法确定对药品安全性和有效性影响的,应设计针对性的比对试验研究,以证实其对药品安全性和有效性是否有影响;(3)对研究显示有差异,评判为不相似的,不宜继续按生物类似药研发。
7.临床研究和评价
7.1一般考虑
临床相似性比对试验研究,应遵循逐步递进的研究原则。通常从PK和/或PD比对试验研究开始,根据相似性评价的需要考虑后续安全有效性比对试验研究。 临床试验用药物应尽可能使用与前期比对试验研究用药物相同批的产品。对不同产地及生产工艺发生改变的,尤其是处方变更的,应重新开展药学或者非临床的比对试验研究。 对前期研究结果证明候选药与参照药之间无差异或差异很小,且临床药理学比对试验研究结果可以预测其临床终点的相似性时,则可用于评判临床相似性。对前期比对试验研究显示存在不确定性的,则应当开展进一步临床安全有效性比对试验研究。
7.2临床药理学
对PK和PD特征差异的比对试验研究,应选择最敏感的人群、参数、剂量、给药途径、检测方法进行设计,并对所需样本量进 行科学论证。应采用参照药的给药途径及剂量,也可以选择更易暴露差异的敏感剂量。应预先对评估PK和PD特征相似性所采用的生物分析方法进行优化选择和方法学验证,使其具备充分的准确度、精密度、特异性、灵敏度及良好的重现性。 应预先设定相似性评判标准,并论证其合理性。
7.2.1药代动力学
在符合伦理的前提下,应选择健康志愿者作为研究人群,也可在参照药适应症范围内选择适当的敏感人群进行研究。 对于半衰期短和免疫原性低的产品,应采用交叉设计以减少个体间的变异性;对于较长半衰期或可能形成抗药抗体的蛋白类产品,应采用平行组设计,并应充分考虑组间的均衡。 对药代动力学呈剂量或时间依赖性,并可导致稳态浓度显着高于根据单次给药数据预测的浓度的,应进行额外的多次给药PK比对试验研究。 对PK比对试验研究,通常采用等效性设计,其界值可使用经典的等效性范围,即80%-125%。对采用其他等效性范围的,应说明理由并论证其合理性。研究中除考察吸收率/生物利用度的相似性外,还应考察消除特征(消除和/或消除半衰期)的相似性。 一般情况下不需进行额外的药物-药物相互作用研究和特殊人群研究等。
7.2.2药效动力学
PD比对试验研究应选择最易于检测出差异的敏感人群和量效曲线中最陡峭部分的剂量进行,通常可在PK/PD研究中考察。对PK特性存在差异,且临床意义尚不清楚的,进行该项研究尤为重要。
对PD指标,应尽可能选择有明确的量效关系,且与药物作用机制和临床终点相关,并能敏感地检测出候选药和参照药之间具有临床意义的差异。
7.2.3 药代动力学/药效动力学
PK/PD比对试验研究结果用于临床相似性评判的,所选择的PK参数和PD指标应与临床相关,应至少有一种PD指标被公认为临床疗效的替代终点,且对剂量/暴露量与该PD指标的关系有充分了解;研究中选择了测定PK/PD特征差异的最敏感的人群、剂量和给药途径,且安全性和免疫原性数据也显示为相似。
7.3有效性
遵循随机、双盲的原则进行比对试验研究,样本量应能满足统计学要求。剂量可选择参照药剂量范围内的一个剂量进行。 对有多个适应症的,应考虑首先选择临床终点易判定的适应症进行。对临床试验终点的指标,应尽可能与参照药注册临床试验所用的一致。对采用其它终点指标的,应经过充分论证。 临床有效性比对试验研究应采用等效性设计。对采用非劣效设计的,应选择合理的非劣效界值,并采用参照药批间或批内的临床疗效差别,评判候选药和参照药之间的相似性。
7.4安全性
安全性比对试验研究应在PK、PD、以及疗效相似性比对试验研究中进行,必要时应对特定的风险设计针对性的安全性比对试验研究。
比对试验研究中,应根据对不良反应发生的类型、严重性和频率等方面的充分了解,选择合适的样本量,并设定适宜的相似性评判标准。一般情况下仅对常见不良反应进行比对试验研究。
7.5免疫原性
应根据非临床免疫原性比对试验研究结果设计开展必要的临床免疫原性比对试验研究。当非临床免疫原性试验研究结果提示相似性时,对提示临床免疫原性有一定的参考意义,可仅开展针对性的临床免疫原性比对试验研究;对非临床比对试验研究结果显示有一定的差异,或者不能提示临床免疫原性应答的,临床免疫原性试验的设计应考虑对所产生的抗体进行交叉反应测定,分析其对安全有效性的影响。 临床免疫原性比对试验研究通常在PK、PD、以及疗效相似性比对试验研究中进行。应选择测定免疫应答差异最敏感的适应症人群和相应的治疗方案进行比对试验研究。当考虑适应症外推时,还应关注不同适应症人群的免疫原性风险,必要时应分别开展不同适应症的免疫原性比对试验研究。 研究中应有足够数量的受试者,并对采样时间、周期、采样容积、样品处理/贮藏以及数据分析所用统计方法等进行论证。抗体检测方法应具有足够的特异性和灵敏度。免疫原性测定的随访时间应根据发生免疫应答的类型(如中和抗体、细胞介导的免疫应答)、预期出现临床反应的时间、停止治疗后免疫应答和临床反应持续的时间及给药持续时间确定。 免疫原性比对试验研究还应考虑对工艺相关杂质抗体的检测,必要时也应开展相应的比对试验研究。
比对试验研究还应对检测出的抗体的免疫学特性及对产品活性的影响进行研究,并设定相似性评判的标准。
7.6适应症外推
对比对试验研究证实临床相似的,可以考虑外推至参照药的其它适应症。 对外推的适应症,应当是临床相关的病理机制和/或有关受体相同,且作用机理以及靶点相同的;研究中,选择了合适的适应症,并对外推适应症的安全性和免疫原性进行了充分的评估。适应症外推需根据产品特点个案化考虑。对于合并用药人群外推至单一用药人群、存在不同推荐剂量的人群之间进行适应症外推时应慎重。
8.说明书
应符合国家相关规定的要求,原则上内容应与参照药相同,包括适应症、用法用量、安全性信息等。当批准的适应症少于参照药时,可省略相关信息。说明书中应描述候选药所开展的临床试验的关键数据。
9.药物警戒
应提供安全性说明和上市后风险管理计划/药物警戒计划,按照国家相关规定开展上市后的评价,包括安全性和免疫原性评价。
10.名词解释
生物类似药:是指在质量、安全性和有效性方面与已获准上市的原研药具有相似性的治疗性生物制品。
候选药:是指按照生物类似药研发和生产的,用于比对试验研究的药物。
参照药:是指已获得国家药品监督管理当局批准上市的,在生物类似药研发过程中与之进行比对研究用的原研产品。
原研产品:是指按照创新药研发和生产并且已获准上市的原始创新性生物制品。
比对试验:是指在同一个试验中比较候选药与参照药差异的试验研究。
来源:医谷网

为你推荐
 资讯
资讯 诺和诺德:司美格鲁肽“保护期”将持续至2027年4月
近日,诺和诺德全球总裁兼首席执行官杜麦克(Mike Doustdar)首度明确表态:司美格鲁肽在中国的监管数据保护将持续至2027年第二季度,届时仿制药才能开始合法进入市场。
2026-06-21 13:42

 资讯
资讯 第四批全国中成药集采与第二批接续采购中选结果
近日,全国中成药联合采购办公室正式发布《关于公布全国中成药采购联盟集中采购中选结果的通知》,备受业界关注的第四批全国中成药集中带量采购及第二批接续采购中选结果尘埃落定。
2026-06-20 21:08
 资讯
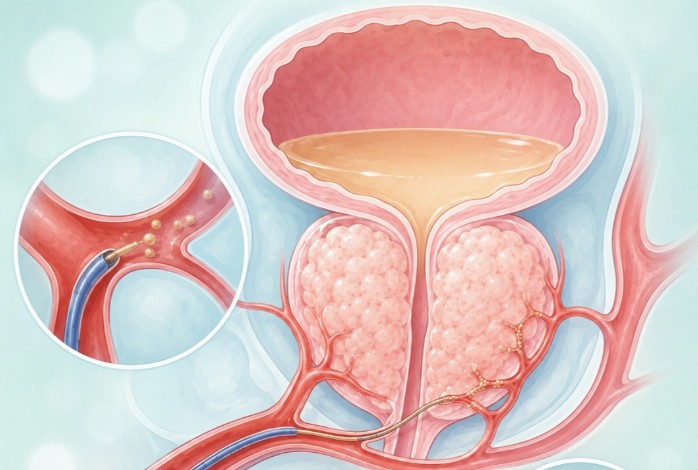
资讯 又是一个父亲节,关注和做好前列腺癌早期筛查
前列腺癌被称为中老年男性的隐形杀手——隐形在于早期几乎没有症状,杀手则在于部分恶性程度高的前列腺癌进展迅速,一旦发生转移,预后显著变差。
2026-06-19 14:59
 资讯
资讯 低活跃预测不等于低风险,台风、飓风、强降雨和洪涝企业应做好哪些应对?
随着北半球夏季极端天气高发期到来,台风、飓风、强降雨和洪涝等风暴相关风险进入重点关注阶段。世界领先的健康和安全风险服务企业国际SOS提醒在全球运营的企业:风暴季准备的关...
2026-06-18 21:41
 资讯
资讯 药石科技发布“绿色智能化学引擎”战略,强化下一代疗法CRDMO平台能力
CPHI China 2026期间,南京药石科技股份有限公司(股票代码:300725 SZ,以下简称“药石科技”)在上海举行集团战略发布会
2026-06-18 13:49

 资讯
资讯 因美纳在华推出移动式生信分析解决方案,推进蛋白质组学生态合作
近日,因美纳正式推出“因美纳生信移动宝”,该生信分析解决方案采用移动式部署模式,旨在将DRAGEN™驱动的高性能算力直接送达科研与临床研究一线。同期,因美纳进一步强化其在...
2026-06-18 09:55
 资讯
资讯 治疗早泄药物国内获批
6月15日,国家药监局药品批准证明文件送达信息显示,Plethora Solutions 和复星医药(600196 SH;02196 HK)联合申报的 5 1 类药物利多卡因丙胺卡因气雾剂获批上市,此为...
2026-06-17 21:36



 资讯
资讯 武田制药公布Oveporexton新关键性研究数据
在第40届美国联合专业睡眠学会年会(SLEEP 2026)上公布的3期研究次要及探索性终点结果进一步显示,Oveporexton在广泛的日间及夜间症状方面带来改善
2026-06-16 12:58
 资讯
资讯 兆科眼科硫酸阿托品滴眼液澳洲上市注册申请获受理
6月15日,兆科眼科发布公告称,公司就其用于减慢儿童近视加深疗法的硫酸阿托品滴眼液(0 02%剂量,产品代码:NVK002)提出的注册申请已获澳洲Therapeutic Goods Administrati...
2026-06-16 10:53
 资讯
资讯 和铂医药和百图生科宣布联合成立AI医药公司
6月15日,和铂医药和百图生科联合宣布,双方将建立全面战略合作伙伴关系,联合创立一家面向全球市场的新型AI管线研发公司MegaStream Techbio。
2026-06-16 10:25
 资讯
资讯 陪审团认定安进故意侵权,和铂医药最高可获6060万美元赔偿
今日,和铂医药(02142 HK)发布公告,美国特拉华州联邦地区法院的陪审团就和铂医药针对安进公司(Amgen Inc )及其子公司Teneobio, Inc (以下合称“安进”)提起的专利侵...
2026-06-15 21:28

 资讯
资讯 年内创新药上市企业最大回购计划
港股上述企业,中国生物制药(正大天晴为其旗下公司)今日发布公告,2026年6月12日,公司董事会决议通过一项股份购买计划,将视市场情况于未来12个月以不超过20亿港元总价在公开...
2026-06-15 15:56
