
因为美国政府停摆,对于FDA的很多关注都留在了FDA会不会因为缺少经费就此关门,但是FDA在危急存亡之秋,依然铁了心推动医疗器械监管的改革,从2018年年底到2019年初始持续密集地发布公告,深入地改革医疗器械审批工作。
其实FDA2018年一整年都在紧抓着医疗器械的安全性问题。从2018年初公布“医疗器械安全计划(Medical Device Safety Action Plan)”;11月,改革510(k)计划;12月4号,更新De Novo审查计划;12月14号又也推出了全新的安全技术计划(STeP);今年1月23号推出510(k)最终指南征集方案。
哪怕在政府关门期间,也没有放弃,而这些政策都只有一个指向,那就是医疗器械的安全问题。这些政策也都将在2019年出台更多的落地细节,可以说2019年是FDA医疗器械监管的分水岭。一向对医疗器械创新大开绿灯的FDA,将全程拉起安全警戒线。
FDA更新De Novo,AI产品审批将更严格
实际上医疗器械的不良反应问题是难以避免的,但是如果产品安全问题、数据安全问题频繁地出现,那么就是监管机构需要反思的问题。
来自ICIJ(国际调查记者协会)报道中的数据显示,在他们调查超过800万条植入医疗器械相关的记录后,得出结论是:在过去10年中,一共有超过540万件不良记录被上报给FDA。直接相关的患者死亡接近8.3万人,造成170多万人受伤。
这是FDA这个守夜人的缺位。但是这一次FDA怒了,开始全面革新。动脉网曾报道过FDA对510(k)革新,接下来将详解FDA将如何更新De Novo和全新推出的安全计划。
De Novo值得受到关注,因为它是很多创新医疗器械上市的出口。比如AI,FDA批准上市的首个IDx的AI糖网筛查就是通过De Novo获批的。
而这次对De Novo的改进主要是明确规则,FDA公布了名为“De Novo分类规定征求意见稿”的文件。如果正式生效之后,将有助于对新型医疗器械进行适当分类。例如,拟议条例中规定了有关De Novo分类的结构、标准、透明度,以及申请及撤回De Novo审批的文件和内容要求的标准。
征求意见稿长达70多页,其中最引人关注的条款是允许FDA对相关医疗设备进行上市前生产检查,作为De Novo审查过程中的一部分。
也就是说FDA将在用De Novo审查医疗器械创新产品中,用到部分严格PMA审查中的标准和流程。
这就需要先理解FDA对于医疗器械审批的重重规则,PMA(上市前审评 Premarket Approval)和新药审评流程差不多,要求进行严格的人体试验,监管风险最高的产品,通常适用于Ⅲ医疗器械;510(k)则是只需要证明需要审评的器械和市场已有的医疗器械类似就可以了。而2014年最新修订的 De Novo则是适用于更多的此前没有同类产品创新医疗器械,同时风险不是很高,无需进行PMA审批。
不同渠道获批的比例大概约为510(k)70%,而PMA则是占到20%左右。这些年来,一共有235种新型医疗设备通过De Novo获得上市批准。
结合到De Novo是FDA批准首个AI产品、首个自适应助听器、首个用于处方治疗的APP等多个创新性数字健康产品上市的渠道。
也就意味很多数字健康公司都要重新审视自己的质量系统,为FDA的审查做好准备。如果说510(k)更多涉及传统医疗器械公司,De Novo规则的改变将波及更多的数字健康公司。
“我们希望做到让De Novo更加高效和透明,通过明确厘清整个审查过程和申请过程。我们也认为会有更多的创新医疗器械产品通过De Novo获得认证,尤其是我们正在采取措施改进510(k)法规的背景下。通过不断地改进De Novo,可以帮助我们更好地监管新的科技,在保证病人的安全的同时,鼓励医疗器械产品创新。”FDA局长Scott Gottlieb博士在声明中表示。
FDA转向,由鼓励创新向鼓励安全创新
正所谓大禹治水堵不如疏,FDA同样为之。在把以前的各个监管口收紧后,它还希望有更多的本身就针对医疗器械安全问题的产品出现,为此,FDA在2018年12月14号推出了全新的STeP(Safer Technologies Program)安全科技计划。
简单来说,STeP释放的信号就是如果有比现有的医疗器械更加安全的产品,也许没有那么创新,FDA也会对它青眼有加,加入STeP计划,在产品研发的前期就可以与FDA的审查专家深入交流,帮助它们更快地获得上市许可。
在2019年,FDA将会公布更多的相关操作细则,进一步详细说明哪些特征适用于STeP。FDA还举了两个例子。例如一种骨科设备,它可能不是用来治疗一种危及生命或者无法逆转的疾病,但是它有一个创新的安全机制,可以减少术后并发症,它就适用STeP。或者一种医疗成像设备,它不符合突破性创新,但是和同类设备相比,它显著减少了辐射暴露,这对患者和医生都大有裨益。与现有设备相比,此类设备具有安全优势,它们就有资格包含在STeP中。
想要更好地理解这个全新的STeP计划,就要先理解FDA现行的Breakthrough Devices Program(突破创新设备计划),Breakthrough Devices Program项目在2016年末期出台,现在已经有110种产品加入该计划,其中有8种实现成功上市。
“突破创新设备计划”是为了鼓励能够让病人得到及时快速的诊断的医疗设备或者能够治疗危及生命的产品,以及能够做到对某些无法逆转的疾病起到作用的设备出现。这些设备要求提供治疗或诊断疾病的新方法,与现有的治疗或诊断相比具有显著优势,其中的典型就是首个帮助判断脑震荡的血液测试,之所以称它为突破器械,是因为这种产品可以减少CT扫描。
前不久,中国大陆抗癌新药获得FDA突破性疗法认定,突破性疗法就是与突破性器械是类似的条例,不过是适用于药物领域。
但是现在FDA认为创新很重要,但是安全必不可少,而且两者应该说是互补的,从Breakthrough Devices Program到STeP计划转向。这也是为什么我们说FDA 2019年对医疗器械的监管重点将是安全的原因之一。
哨兵计划,FDA监管方法的分水岭
而另一个原因则是体现在FDA的“五年计划”中。从点到面,FDA都在强调安全性的监管。其中主要的体现就是FDA今年1月公布的“哨兵系统”(Sentinel system)。
FDA推出的“哨兵系统”和FDA-Catalyst这个FDA布局了近10年的计划,正在随着信息化、大数据的发展开始正式浮出水面。
“哨兵系统”是FDA用来大规模电子安全监控的工具:在保护个人医疗记录隐私的同时,能够获取和分析数百万患者使用的医疗产品的体验数据。
如果说十多年前,建立一个如此大规模的电子监控系统还只能是某一位富有远见的领导人的想法;那么今天,它已经变成一个监控FDA批准的药品和其他所有医疗产品安全性的国家电子系统,这个系统命名为“哨兵系统”(Sentinel system)。
“‘哨兵系统’的发展可以说是FDA监管的分水岭,我们现在还在寻找更多的方法来构筑这个坚实的系统,开发更好的工具使用数据来提高安全性。”Scott Gottlieb表示。
“哨兵系统”不仅是FDA安全性监控的重要工具,它也是FDA方法论创新的关键引擎,它也是推进真实世界研究(RWE)的重要平台。
在实施“哨兵系统”之前,FDA对于医疗产品的安全监测的数据主要来自于患者、医护人员、制药行业以及其他行业的不良事件报告。这些报告是FDA不良事件报告系统(FDA’s Adverse Event Reporting System)中“被动监测”系统的一部分,对安全研究人员来说仍然具有重要意义。
但是,“哨兵系统”的“主动监视”功能是对FAERS数据极其重要的补充。它不需要等待安全数据,而是让FDA在需要的时候直接获取数据。
事实上,FDA的不良事件报告系统一直被认为是摆设。FDA有上报不良事件和并发症的系统,但是上报数量严重不足。在这个自行上报的系统中,医生如果观察到不良事件,没有上报的义务,而生产商有上报的义务,却是最不愿意上报的人。
也就导致了某些医疗器械产品在植入几个月甚至多年以后,才被发现有严重的副作用,但是那时候已经有成千上万人植入了。例如最近受到调查的美敦力和波士顿科学的涂有紫杉醇的某些支架。
2018年12月发表于美国心脏协会杂志的研究发现,在膝盖上使用涂有紫杉醇涂层设备两年后,死亡风险意外增加。而风险被爆出之后,FDA才紧急启动调查。
“哨兵系统”从2009开始试点,2016开始全面推行。目前“哨兵系统”的数据来源主要依赖保险索赔数据。接下来FDA希望接入更多的EHR数据到“哨兵系统”的上市前和上市后监控中。FDA还在开发知识管理系统,并且利用AI推进自然语言处理,以评估索赔数据和EHRs中的信息。
在医疗创新中,创新一直被当做行业重要的驱动力。但是从FDA的一系列举措来看,FDA在2019年更看中医疗器械的安全保证。更严格的监管正在逐渐上线,当然这也不止于医疗器械,而是有关所有的医疗产品。
这种用数据进行监管的方法也大大减少了人为的错漏。消除腐败滋生的土壤。ICIJ对美国司法部(Justice Department)和美国证券交易委员会(Securities and Exchange Commission,简称sec)的数据进行的一项审查显示,自2008年以来,美国和其他国家的器械制造商已经支付了至少16亿美元,用来就腐败、欺诈和其他违规行为与监管机构达成和解。
在今年,我们肯定还能看到更多关于FDA加强医疗器械安全性监管的动作,其中FDA最的值得借鉴的就是如何平衡医疗设备的创新和安全问题。在不断地涌入创新概念和产品的环境下,调整监管机构的监管框架。
国内药监局拉紧高压线防止踩雷医疗器械
而对于国内来说,我国的器械审评审批中心一向被誉为是全世界最严格的审评中心。而在上市后的监测中,卫健委和国家市场监督管理总局在2018年8月13日共同发布了《医疗器械不良事件监测和再评价管理办法》 ,自2019年1月1日起施行。其中明确了医疗器械不良事件监测的两个主体:一个是医疗器械上市许可持有人,另一个是国家药品监督管理局建立的国家医疗器械不良事件监测信息系统。
在上市后的监测方面和FDA的方法相差无几,也是由制造商主动担任起监测的责任,主动上报到监测系统中。
药监局同FDA一样也在探索如何平衡创新和安全。
早在2014年,原国家食品药品监督管理总局就发布了《创新医疗器械特别审批程序(试行)》文件,2015年,《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)和《中央办公厅国务院办公厅关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)持续出台。2018年1月,药监局重磅推出了《创新医疗器械特别审批程序》。
截止2018年12月31日,已有197个产品进入创新医疗器械特别审查通道,批准神经外科手术导航定位系统、正电子发射断层扫描及磁共振成像系统等54个产品注册,一批创新性强、技术含量高、临床需求迫切的创新产品上市,填补了相关领域的空白,更好的满足了人民群众的健康需求。
不得不为药监局鼓掌的是,在为创新医疗器械开路的同时,也把创新医疗器械的安全性作为重点监测对象。2018年8月13日公布的《医疗器械不良事件监测和再评价管理办法》中第四章提到:
第四十七条 创新医疗器械持有人应当加强对创新医疗器械的主动监测,制定产品监测计划,主动收集相关不良事件报告和产品投诉信息,并开展调查、分析、评价。创新医疗器械持有人应当在首个注册周期内,每半年向国家监测机构提交产品不良事件监测分析评价汇总报告。国家监测机构发现医疗器械可能存在严重缺陷的信息,应当及时报国家药品监督管理局。
在具体的实施细节上,国内和国外有不同的方式,也各有所长。但是FDA利用大数据、AI等技术进行监管方法论创新这一点上,值得我们思考和借鉴。
来源:动脉网
作者:杨雪
来源:动脉网 作者:杨雪

为你推荐
 资讯
资讯 恒瑞医药双靶点GLP-1/GIP瑞普泊肽注射液中国T2DM口服药控制不佳人群Ⅲ期研究取得积极顶线结果
近日,恒瑞医药宣布,GLP-1 GIP双重受体激动剂瑞普泊肽(HRS9531,大中华区外称KAI-9531)注射液治疗经二甲双胍单药或联合SGLT2抑制剂治疗后血糖控制不佳成人2型糖尿病患者的中...
2026-08-07 17:23
 资讯
资讯 韩宇药业提交了替尔泊肽的国内首仿上市申请
据国家药审中心官网显示,8月6日,翰宇药业申报的替尔泊肽注射液上市申请获得受理,注册分类为4类(境内外均已上市的药品的仿制药)。从目前公开信息看,这是国内首个替尔泊肽注...
2026-08-07 16:35
 资讯
资讯 礼来半年报业绩大增,二次上调全年业绩指引
8月5日,礼来公布了2026年上半年业绩——总营收427 73亿美元,同比增长49%。其中,第二季度营收229 74亿美元,同比大增48%。此前,礼来第一季度实现营收197 99亿美元,同比增...
2026-08-07 15:57
 资讯
资讯 全球首款 mRNA 季节性流感疫苗诞生!Moderna mFLUSIVA 获 FDA 批准
美国食品药品监督管理局(FDA)正式批准 Moderna 研发的 mRNA 季节性流感疫苗mFLUSIVA(mRNA-1010)上市,用于 50 岁及以上成人预防季节性流感
2026-08-07 13:07
 资讯
资讯 荣昌生物2026年半年度业绩
根据预告,荣昌生物预计 2026 年半年度实现营业收入约 58 5亿元,与上年同期相比,将增加收入约 47 52亿元,同比增加约 433%。预计 2026 年半年度实现归属于母公司所有...
2026-08-06 21:31

 资讯
资讯 信达生物2026上半年产品收入超2024年全年
公告显示:2026年上半年,信达生物产品收入实现超人民币82亿元,同比增长55%以上。其中,公司第二季度产品收入录得超过人民币43亿元,同比增长约60%。根据信达生物此前发布的信...
2026-08-06 15:09
 资讯
资讯 百济神州2026上半年业绩大增,上调全年业绩指引
2026年半年度公司营业总收入222 20亿元,较上年同比上升26 8%;2026年半年度归属于母公司所有者的净利润32 71亿元,较上年同比上升627 1%。
2026-08-06 14:14
 资讯
资讯 因美纳MiSeq i100 Series-CN中国上市,以全球品质与本土制造支持中国客户测序能力建设
MiSeqTM i100 Series-CN正式在中国上市,首批由因美纳中国生产制造基地生产的本土化产品已于近期完成客户交付。
2026-08-06 13:33
 资讯
资讯 石药集团与阿斯利康成立合资公司,收到1000万美元里程碑付款
双方将在河北石家庄建设新一代生物制剂生产基地,该合资公司的初始业务将聚焦于为全球市场生产及供应双方约定的生物制剂药物原液(DS)。
2026-08-05 20:49
 资讯
资讯 全球首款!核心医疗人工心脏产品获欧盟CE认证
Corheart®6 可用于桥接心脏移植、短期心肌功能恢复治疗以及长期终点治疗,完整覆盖终末期心衰从短期生命支持到永久循环辅助的全场景临床需求
2026-08-05 20:37
 资讯
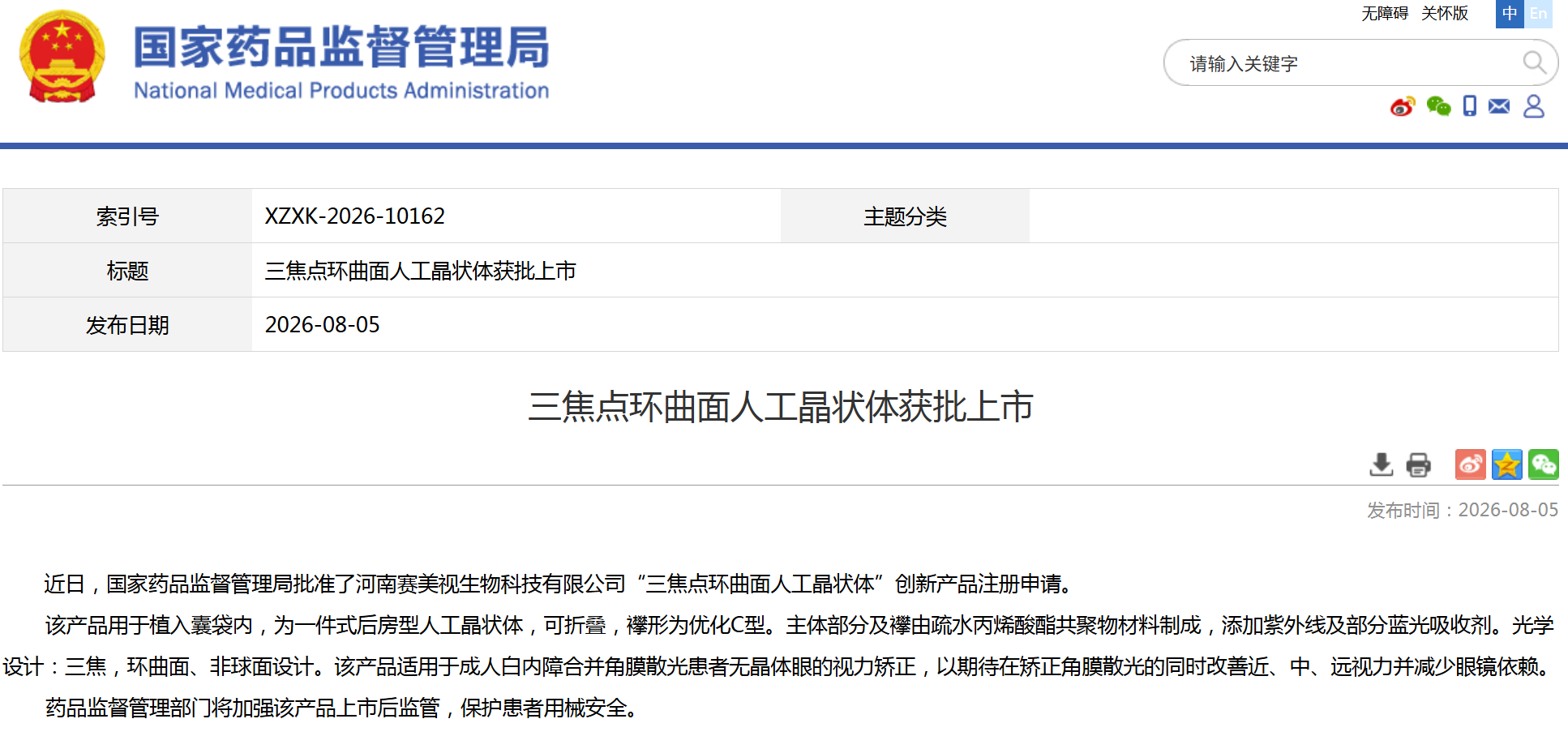
资讯 国家药监局批准3款创新医疗器械上市
近日,国家药品监督管理局批准了河南赛美视生物科技有限公司的“三焦点环曲面人工晶状体”和深圳北芯医疗科技有限公司的“心脏脉冲电场消融仪”、“一次性使用心脏脉冲电场消融...
2026-08-05 16:47
 资讯
资讯 80亿美元,全球两家核药公司合并
美东时间8月3日,Curium Pharma宣布与Lantheus Holdings, Inc 达成最终合并协议。Curium将以每股102 5美元现金收购Lantheus全部流通股,基础对价70亿美元,若2030年前达成...
2026-08-05 13:51
 资讯
资讯 章科:坚持为人民出政绩 推动医疗保障事业高质量发展
医疗保障制度依法保障每一位参保群众的医保权益,有力提高全社会医疗服务的可及性,有助于增进民生福祉、缩小贫富差距,促进社会公平和共同富裕。
2026-08-05 10:48
 资讯
资讯 康宁杰瑞与PathosAI达成22亿美元ADC授权协议,1.25亿美元首付款
8月4日,康宁杰瑞生物制药宣布其全资子公司江苏康宁杰瑞生物制药有限公司与美国临床阶段生物技术公司Pathos AI就TROP2 HER3双特异性抗体偶联药物JSKN016达成独家授权许可协议。
2026-08-04 22:14
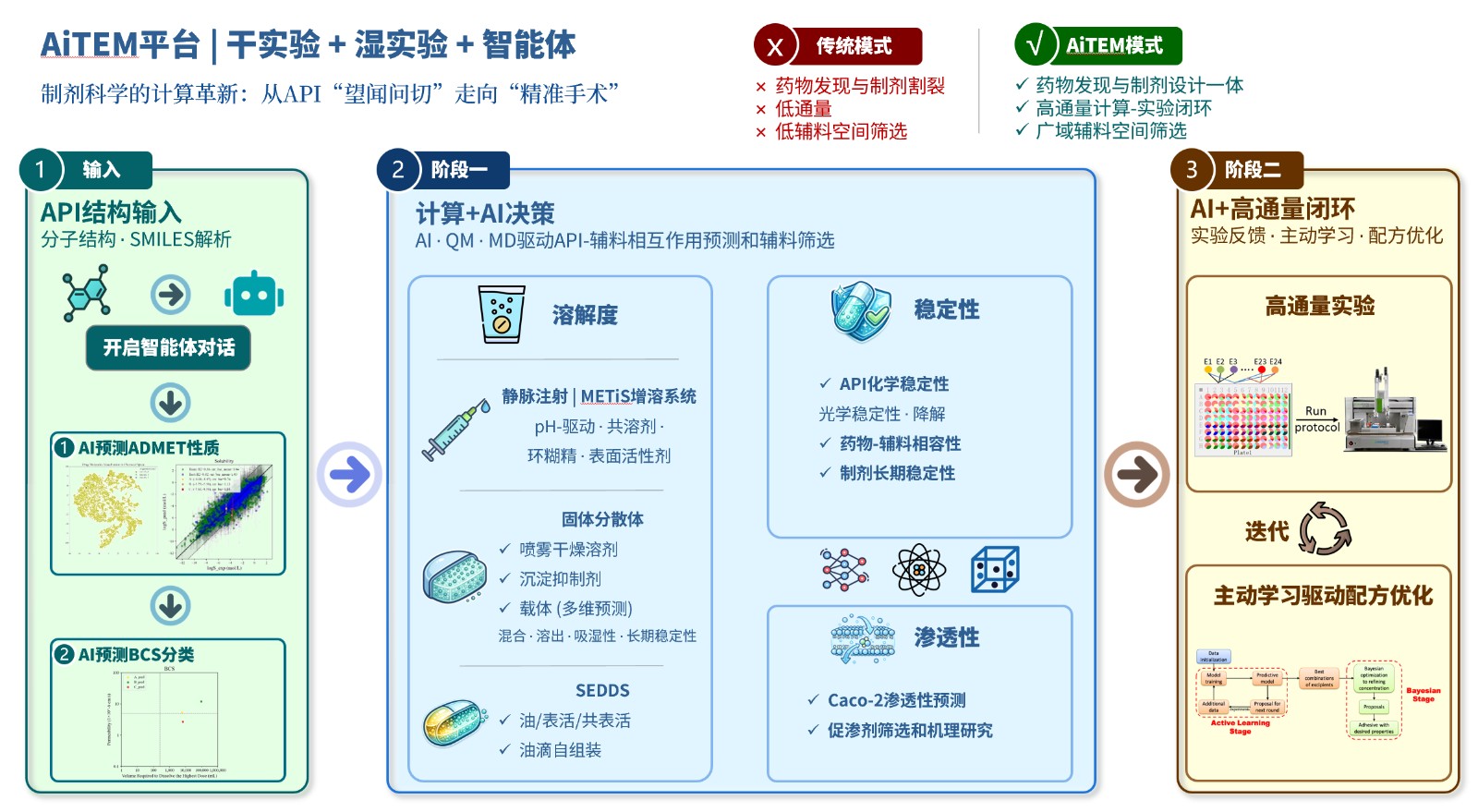 资讯
资讯 剂泰科技与恒瑞医药达成AI制剂合作
8月3日,剂泰科技发布自愿公告,宣布自主研发的AiTEM人工智能制剂创新平台将在恒瑞医药进行本地化部署,加速药物制剂配方开发效率。
2026-08-04 13:01
