
9月19-22日,备受关注的第22届全国临床肿瘤学大会暨2019年CSCO学术年会在厦门国际会展中心如期召开。假如评选一下谁是CSCO上最受关注的讲者,恐怕众星云集的肿瘤临床泰斗、大咖们都需要让位于我们肿瘤新药上市的“守门人”——CDE(药品审评中心)。
如往年一样,今年依旧是CDE化药临床一部部长杨志敏率领她的团队代表监管部门与制药界、医学界的同仁们汇报成果、交流心得、解答疑问。
无论是19日下午有临床医生、制药企业、CRO公司参与的“2020中国抗肿瘤创新药升级之路”专场,还是21日上午在海峡厅进行的“CDE/CSCO专场——研发审评并举,推进抗肿瘤新药上市”都座无虚席,甚至大会临近尾声时,由杨志敏部长参与主持的“CSCO临床研究专家委员会——新药研发专场”依旧是被围堵得水泄不通。
为什么由CDE参与的会场都如此火爆?
监管机构努力重塑行业信任
无论在哪个国家,可能都没有比食药监管机构更受挑战的政府部门了。比如上世纪50年代的EMA,在沙利度胺大规模致残致畸事件发生时,信任感也是迅速降到了冰点。
在我国,随着疫苗以及部分药品重大安全事故的发生、仿制药劣币驱逐良币的现象,亦是使民众和业界对监管机构的信任快速下滑。甚至很多行业人士将21世纪初我国创新药发展受制归咎于政府的不作为。一句话说,当产业发展受到制受约,从业者第一反应便是监管部门的“锅”。
摧毁信任只要一瞬间,重塑却可能得上百年。近年来CDE多次走近行业,与一线交流,传递的讯号无外乎是监管机构是支持产业发展、支持创新的讯号。
CDE/CSCO专场不容置疑是这届CSCO的重中之重,以下内容主要是根据“CDE/CSCO专场”传递的一些讯息做了整理。
首先这场报告准备了足足两个月。杨志敏部长说,两个月前,CSCO秘书长、军事医学科学院附属医院江泽飞教授就专程来到CDE,与她探讨CDE/CSCO上的主要汇报内容。这也不难看出,CSCO和CDE对这场“官民互动对话”的重视。最后,CSCO主办方和CDE将这次的专场报告内容确定为5个方面:
杨志敏部长汇报《2019年中国抗肿瘤新药审评情况报告》
CDE高级审评员周明主题报告《抗肿瘤小分子创新药临床研发与审评考量》
CDE高级审评员张虹主题报告《抗PD-1/PD-L1肿瘤免疫治疗临床研发与审评考量》
CDE高级审评员夏琳主题报告《抗肿瘤生物类似物临床研发与审评考量》
CDE统计与临床药理部王俊主题报告《真实世界数据用于抗肿瘤新药注册的审评考量》
本文主要以杨志敏部长演讲内容的核心要点来看监管部门做出的努力。
(1)2019年抗肿瘤药物IND申报情况
小分子药物靶点分布相对均匀,热门靶点包括CDK4/6、EGFR-T790M、PARP、BTK、AKT、HADC、MET、NTRK、FGFR等。
大分子以单抗为主,ADC、双特异性抗体显着增加,免疫治疗药物活跃、PD-1/PD-L1扎推。
截止目前,一共暂停了28项临床试验,原因有非临床、临床以及药学这三大方面。非临床的原因主要包括:药效学不充分(未体现抗肿瘤量效关系)、存在重大不可控的安全性问题(严重心脏毒性、肝毒性);临床的原因主要包括:方案设计有重大缺陷;例如起始剂量依据不充分,全新两药联合无药效学依据;药学的原因主要包括药学质量不可控;例如非临床和临床的工艺重大变更,未开展可比性研究。
(2)2019年抗肿瘤NDA申报情况
NDA注册申请逐年增多,其中抗肿瘤药物占的比例大约为29%。
1类创新药的NDA申报数量逐年增多,2018年高达47个,截止到2019年8月已经有29个进行申报,其中抗肿瘤占比大约46.5%;申报适应症类型最多的是非小细胞肺癌,其次是乳腺癌和非霍奇金淋巴瘤。
2018年抗肿瘤化药和生物制品NDA审评通过情况达到60%,不批准主要原因来自临床和药学两大方面。临床原因包括:未达到预设标准;达到统计学意义、未达到临床意义(例如:PFS延长21天);人群混杂,同时包括多线患者,未分组或者是两组不均衡;药学原因主要是未采用上市工艺和规模,且无可比性研究数据。
(3)抗肿瘤药开发的审评考量
建议与CDE的沟通:包括Pre-IND、试验过程中以及Pre-NDA的沟通。CDE对沟通交流会议回复以书面回复为主要回复形式。
沟通的焦点问题主要集中在3个方面,一是早期临床研究阶段,二是联合用药,三是关键临床阶段。
早期研究临床评价:考量主要包括研发立题合理性(科学基础),即:非临床机制研究是否支持,毒理研究是否充分以及同靶点药物的研发进展;方案设计科学性(FIH),即:起始剂量是否合理,爬坡设计是否恰当;研究人群是否符合当下临床试验并排除了高危风险患者;DLT定义是否充分,有无明确的爬坡停止条件;安全性监控是否充分?有无完美的风险控制计划。
在提到国内外同步早期研究时,需要考量种族差异,包括疾病背景的差异(病因及流行病学)、临床医疗实践的差异(背景及对照的选择)以及剂量的合理性(耐受性和代谢)。当前对国内外同步早期研究的考虑主要分为参照ICH E17进行平行毒理I期临床试验;国外已获得充分的数据后,可以参照 ICH E5进行简化I期临床试验。
联合用药审评考量:以2种药物的联合用药为例,主要分为3类,分别是A、B均未上市;A上市、B未上市;A、B均未上市。对于联合用药的单药,第一步是得具备联合用药的合理性;第二步是须获得单药的人体药代动力学特征、安全性特征、单药的安全剂量范围,并获得相对明确的II期推荐剂量(RP2D),第三步包括初步获得单药在联合方案拟定目标人群中的量效关系和有效性数据。
进入关键研究临床评价考量:临床定位。首先是找到产品的特点,尤其是区别于其他产品的特点,比如其作用机制、有效性、安全性、依从性的优势是什么?在考虑是单臂设计,还是优效设计,还是非劣设计来证明优势。
前期安全性和有效性数据累积是进入关键研究临床评价标准。一般来说,I期临床剂量递增试验需要纳入20-30例受试者,目的是获得单次和多次耐受性;单次和多次药代以及推荐II期剂量。II期临床是剂量扩展试验,建议进行创新性试验设计。对于单臂或随机对照试验小样本,单臂试验至少要有30例有效性数据,目的是初步疗效和安全性观察、剂量-效应探索以及确定III期剂量和研究人群。
确定人群:是在全人群试验还是开展生物标志物细分人群?是进行大瘤种试验还是在罕见瘤种中开展临床?是直接在一线使用还是先后线使用?
确定给药剂量及方案:给药剂量一般可根据药效学特征,比如受体占有率;可根据药代动力学特征,比如线性或非线性关系;可根据剂量-安全性关系,如DLT及MTD;可根据剂量-疗效关系,如谷浓度/峰浓度与疗效关系;还可按照体重、体表面积给药或者是固定剂量。给药方案一般分为BID或QD。有时候还会考虑单药还是联合用药(与标准相比)。杨志敏还谈到,鼓励建模和模型的应用。建模一定要合理,不是套用论文文献的其它药物模型。
GO or NO GO的决策:首先要不忘初心,是否达到了预期?即临床期望和同类药的数据;其次要审时度势,前面已有多个新药进入关键研究,本品的优势是什么?最后是要忍痛割爱。假如暴露的安全性信息与同类药物相比,具有重大缺陷,需要断舍离,及时止损。
除了以上几个方面,杨志敏部长还提到关键研究临床评价考量需要考虑方案设计、独立影像评估IRC、IDMC职责、CMC挑战以及是否要考虑伴随诊断。CDE的态度是鼓励药物与伴随诊断产品同期申报,同期批准。
(4)NDA申报及纳入优先审评原则
NDA申报,单臂试验要求IRC评估的ORR95%的置信区间下限需达到预设目标值,同时结合缓解持续时间判断,可滚动提交数据;随机对照研究可接受期中分析,但必须为方案和SAP预设。
纳入优先审评的原则,首先是临床急需且具有突出的临床价值,针对NDA申请基本可以确定(尽管pre-NDA沟通决定);目前可接受的标准有:一是目前尚无标准治疗的适应症领域现有的有效治疗;二是与现有治疗相比,患者可显着获益(例如:HR<0.5,PFS延长一倍以上)。
2019年获批的优先审评品种中,除了达可替尼片一线治疗NSCLC的HR略大于0.5(但具有显着OS获益),其他5个品种的HR皆小于0.5,3个品种的单臂研究客观缓解率都显着优于历史对照数据,1个品种为首个生物类似药申请上市。
总的来说,我国创新肿瘤药的挑战包括:基础研究是短板,转化能力不足(创新能力);多学科知识的整合与合作不足(多学科合作);深入分析数据的研究能力有待提升(从失败中成长);科学工具的应用能力还需加强(科学工具助力研发);以临床为目标的差异化研发还需扩展(以患者需求为目标);急功近利的驱动带来更多不确定性(蹄疾步稳)。
杨志敏部长从宏观角度讲述了肿瘤创新药开发取得的成效,与产业界沟通了如何进行方案设计、早期临床研究实验、关键性临床研究等问题。在她的演讲之后,4位CDE审评技术人员又分别从小分子、PD1/PD-L1、生物类似物、真实世界研究等方向谈到了肿瘤药物的临床研发与审评考量。
行业从业者水平参差不齐
在随后的审评员报告中,参会者最关注的的是《抗肿瘤小分子创新药临床研发与审评考量》和《抗PD-1/PD-L1肿瘤免疫治疗临床研发与审评考量》以及《抗肿瘤生物类似物临床研发与审评考量》。放在最后一个演讲的《真实世界数据用于抗肿瘤新药注册的审评考量》可能与受众精力耗尽有关,现场互动的人数明显没有前3个报告多。
参加CDE专场的人员中,80%是药企人员,而且可能有不少是带着领导布置的任务而来。只不过,审评员在演讲时提到的一些特殊情况的解决方案以及背后的考量逻辑,在提问环节仍然会被观众问到,甚至有个别问题是前一位提问者问到,后一位提问者又问到相似问题,实质上背后的逻辑和思路完全一致。
针对这种情况,CDE的代表们仍然是笑脸相迎、耐心解答,无处不彰显对医药从业者是“满满的爱”。仔细想想,这还只是在台上,抛给审评员们的问题就已经应接不暇。如果在日常审评工作中,审评员面临以及需要解答的问题就更加琐碎繁重了。
药审机构一年需要召开几百场的沟通会,当一个人每天面临巨大的审评工作和任务之余,还得回答来自业界实质上已经写进了各类指南的标准问题。其中得需要多大的耐心?这也使得监管部门难免感叹,希望产业界能够换位思考,提问时不要具体到某个问题,而是针对共性问题讨论。
笔者前不久采访一位曾在CDE工作十载有余的前辈,他感叹道:“整个产业需要再上一个台阶,人才培养任重道远,因为很多人的认知意识、学习能力还停留在上个世纪。”
投资者希望通过政策红利快速变现
如果说CSCO的初衷是学术会议,而如今它还被赋予了商业和投资的属性。从参会人员的分布便不难看出。
最早的时候,CSCO是一个学术性质的交流会议,参会者主要是临床肿瘤医生和药企研发人员;后来慢慢地,CSCO的商业属性逐渐增强,大量的药企医药代表、商务BD、市场人员会加入;今年,参会的投资机构人员似乎远超过往年。
“今年参会的投资机构好多噢,除了我,感觉坐在前后左右的参会者都是券商……”不止一位参会者像笔者吐槽,“尤其是某个药品的临床数据一发布,他们能立马关联到相应的上市公司,发雪球,微观股价变化……唧唧咋咋的。”
“我的感受是这群券商真的很拼,会议PPT实时拍摄,一个演讲嘉宾的演讲结束了,立马就转成了PDF文件或者有道云笔记,在各个群里传播着……”另一位参会者笑言,按照这个趋势,随着5G的发展,如果短视频、直播也大量渗透到医药行业,今后可能都不用来现场参会了。
“没办法,很正常,华尔街的投资者们也是这样。像我们做对冲基金的,拼的就是信息不对称。其实中国医药行业目前真正有价值的医药公司也就那么几家,一双手数的过来……”一位由产业转投资的小伙伴向笔者小心透露,“我们是做投资的,低买高卖,赚个差价,确实比之前做药来钱快多了……不瞒你说,之前在制药界倒瓶子付出的十年青春,行情好的话,做投资半年就赚回来了。”
“你也是学生物的,咱当年谁不是因为一句「21世纪是生物学世纪入坑的」,一鼓作气读到了PHD?”一位在业界驰骋20余年的老兵向笔者反问道,“现在回过头来看,生物的应用不外乎去做药,要么去做诊断试剂。”
“从专业难度上讲,做药更有挑战性。于是大多数人从「科研民工」摇身一变「制药民工」。”他继续娓娓道来,“但做了很多年后到头来发现,医药行业往高大上说是「以患者为中心」,而事实上还是一个to B端的行业,产品好不好的话语权并不在终端用户,主要是依赖临床医生手中的那支笔。比较惨的情况,可能连监管机构的那支笔都通不过……”
“CSCO是很好接触医生和监管机构的机会,作为投资机构,我们也想听听客户的声音,才能更好地指导我们投资。如果我们投的产品,根本不符合监管机构的初衷,以至可能根本无缘上市,那才是真正的亏大了……”一位投资者向笔者说明了他不做二级市场,但作为一级投资者也来参会的原因。
来源:医药魔方 作者:玉见

为你推荐
 资讯
资讯 中国医药工业主营业务收入100强
近日,由中国医药工业信息中心举办的2026第43届全国医药工业信息年会在北京开幕,备受关注的“2025年度中国医药工业主营业务收入前100位企业”同步揭晓。
2026-07-14 19:55
 资讯
资讯 国家药监局批准1类创新药阿更葡糖钠注射液上市
近日,国家药品监督管理局批准杭州奥默医药股份有限公司申报的1类创新药阿更葡糖钠注射液(商品名称:奥美克松)上市,该药品用于拮抗罗库溴铵诱导的神经肌肉阻滞。该药品上市为...
2026-07-14 17:01
 资讯
资讯 迪哲医药最高15亿美元BD阿斯利康
今日,迪哲医药发布公告,公司授予阿斯利康在全球范围内的独家开发、商业化舒沃哲的权利。公司将获得阿斯利康支付的一次性、不可返还的首付款6亿美元,最高达4亿美元的临床开发...
2026-07-14 14:57
 资讯
资讯 国家医保局、国家药监局开展跨部门联合检查
近日,国家医保局、国家药监局组成联合检查组,依据药品追溯码筛查线索对内蒙古自治区和山西省部分地市医药机构、药品批发企业开展联合检查。国家医保局党组成员、副局长黄华...
2026-07-14 11:46
 资讯
资讯 2026年国家医保目录及商保目录调整通过形式审查名单公布
2026年目录调整申报阶段,国家医保局共收到基本医保目录申报信息800份,涉及药品通用名664个,最终601个通过形式审查,其中目录外368个,目录内233个,总体通过率91%。共收到商...
2026-07-14 10:34


 资讯
资讯 全国首笔药品追溯码无追索权保理业务在温州落地
近日,在温州市医保局的积极推动下,全国首笔基于药品追溯码的无追索权保理业务成功落地。国药控股温州有限公司凭借药品追溯码形成的真实交易记录,将其对温州医科大学附属眼视...
2026-07-13 10:15

 资讯
资讯 时隔8年调整的2026版国家基药目录新增的4种1类创新药和独家品种
7月9日,国家卫生健康委、国家中医药局、国家疾控局三部门公布2026年版国家基本药物目录。国家基本药物目录在新医改启动后,于2009、2012、2018年完成三轮更新,这是时隔8年后的...
2026-07-12 21:31
 资讯
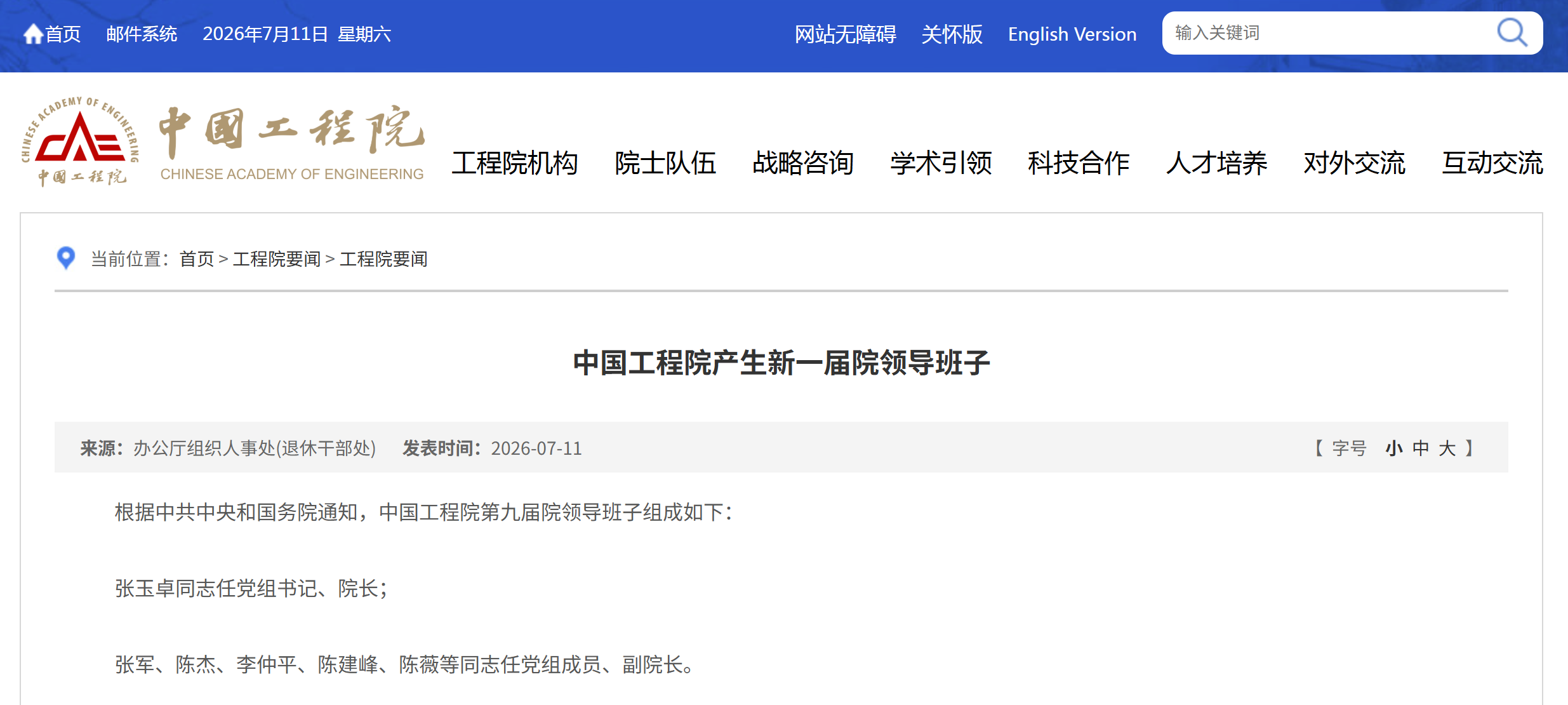
资讯 陈薇院士出任中国工程院党组成员、副院长
据中国工程院官网消息,根据中共中央和国务院通知,中国工程院第九届院领导班子组成如下:张玉卓同志任党组书记、院长;张军、陈杰、李仲平、陈建峰、陈薇等同志任党组成员、副...
2026-07-11 21:36

 资讯
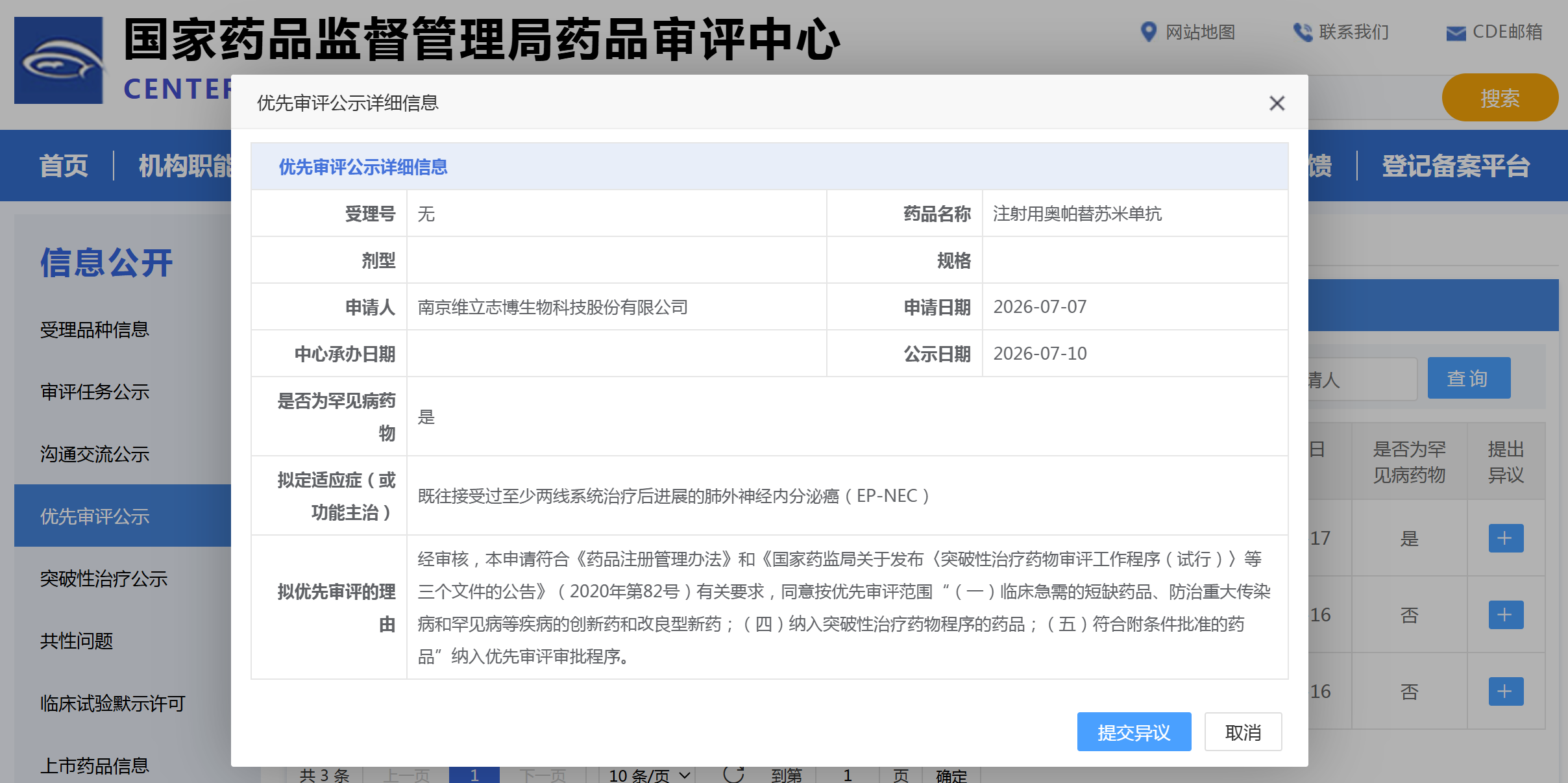
资讯 维立志博PD-L1/4-1BB 双抗拟纳入优先审评
7月10日,国家药监局药品审评中心官网显示,南京维立志博生物自主研发的注射用奥帕替苏米单抗拟纳入优先审评品种,申报适应症为既往接受过至少两线系统治疗后进展的肺外神经内分...
2026-07-10 15:08
 资讯
资讯 丰原药业实控人拟变更为安徽蚌埠市国资委
7月9日,丰原药业发布公告,公司控股股东安徽丰原集团有限公司的一致行动人7月8日与蚌埠投资集团有限公司签署《股份转让意向协议》,蚌埠投资集团拟受让丰原集团的一致行动人持...
2026-07-10 13:49
 资讯
资讯 这家药企上半年净利润增长超5倍
7月8日晚间,海思科发布2026年半年度业绩预告,预计报告期内净利润为7 9亿元-8 7亿元,同比增长513 25%-575 35%。
2026-07-09 19:34
 资讯
资讯 德睿智药5200 万美元 B 轮融资落地,AI 设计口服 GLP-1 冲刺商业化
本轮融资由泰珑投资、申银万国投资、谢诺投资、星河湾集团联合领投,多家头部人民币、美元基金跟投,凯乘资本担任本次融资独家财务顾问。
2026-07-09 14:49
 资讯
资讯 时隔8年,2026年版国家基本药物目录发布
7月9日,国家卫生健康委、国家中医药局、国家疾控局三部门公布2026年版国家基本药物目录。国家基本药物目录在新医改启动后,于2009、2012、2018年完成三轮更新,这是时隔8年后的...
2026-07-09 12:43