
自2017年原国家食药监总局发布《细胞治疗产品研究与评价技术指导原则》,2018年3月首个CAR-T产品的IND申请获批以来,中国的细胞治疗产品的开发与申报可以说进入了全新的阶段。
据药品审评中心(CDE)生物制品临床部部长高晨燕在近日天津举办的中国肿瘤免疫治疗会议上介绍,截至目前,CDE已经受理49个细胞治疗的IND申报。其中CAR-T产品32个,干细胞产品8个。获得临床许可的为19个,其中CAR-T 产品13个,TCR-T产品1个,干细胞产品3个,其他细胞产品2个。
细胞治疗领域的发展如此快速,在产品的开发、申报与评价上难免面临着一些挑战。来自CDE的多位资深审评专家在大会上结合近两年来受理的细胞治疗申报的具体情况,从药学、临床前、临床等多方面、全方位解读一些常见的共性问题、注意事项与更规范的临床试验方案。其中,全程质量管理、风险控制、思路转变与有效的沟通交流是关键。
两大差异与必要的门槛
有关目前中国细胞治疗产品的申报情况,有两大差异已经引起了药监部门的关注。
一是以药物路径申报临床试验(IND)与Clinicaltrials.gov上登记差异很大,在Clinicaltrials.gov上搜索,中国有210个临床试验注册,美国为116个,而如前介绍,真正按照药物IND申报的为49个;第二是IND申请与获得批准的差异。据高晨燕部长介绍,除了正在审评和待申请人补充资料外,18个受理号被暂停临床试验,6个主动撤回。获批与暂停的比例接近1比1。
谈到IND申报数量与Clinicaltrials.gov上登记数量的差异,高晨燕表示,由研究者发起、医院伦理委员会批准的临床试验占有的比例很大。针对IND申报数量少,有人认为是按药品申报门槛较高,导致企业不敢来报,批准数量也较少。
“对于任何在人体进行的临床试验,都会有一定的门槛。我们设置门槛的目的就是要保证受试者的安全,保证细胞治疗产品的质量。细胞治疗产品有一个明确的体外处理过程,而最终产品又不能通过终端灭菌、过滤等方法来控制,因此生产过程的控制非常重要。目前在医院制剂中,大输液用的葡萄糖注射液、生理盐水注射液都被禁止在医院制备,相比之下细胞治疗产品的制备过程风险更高,无法仅通过最终产品进行控制,所以在细胞制备、生产和应用上必须要有门槛。我们希望细胞治疗产品的临床试验应该首先能够保证受试者安全,不要增加除细胞特性以外的额外风险。”
而一半的IND申报暂时未获批准,其中95%的原因是药学研究方面存在问题,药理毒理则占20%,临床试验方案则均存在或多或少的问题,虽然不会直接导致申请驳回。
对于批准临床试验门槛高的质疑,高晨燕特别回应,药监局的技术审评是基于临床试验方案的评价,根据目标受试者的情况,评价细胞治疗产品可能给受试者带来的风险,制定阶段性的要求,不会按上市的标准评价IND申请。阶段性要求中,最重要的还是保证受试者在临床试验中的安全。
全程质量控制,可执行的风险把控
事实上,细胞治疗的高风险性,让质量的全程控制与风险的严格把控成为药监部门考察的重中之重。
CDE生物制品药学部一位审评员表示,曾有很多企业提问,国外已经批准上市产品的放行质量标准能不能拿来直接用。答案是不行的。
因为质量控制是一个整体,一个系统。产品的质量控制不仅是最终产品的放行标准,还包括生产过程中的控制、放行检测控制以及临床使用前的质量核准。国外已上市产品在生产过程中可能进行了较为全面的生产过程控制,所以放行标准是可以进行一些简化,而其他企业产品在过程中可能缺乏全面合理的过程控制的情况下,简单的仿照标准是不合理的。
而细胞治疗产品的质量研究和工艺控制水平等,也会影响细胞治疗的临床疗效和安全性。例如,据CDE观察,在部分CAR-T细胞产品的非注册临床研究中,个别患者回输后当天即出现高热、感染,甚至继发呼吸加快、血压下降、器官功能障碍等休克表现,不良事件的发生规律和实验室检查指标与细胞因子释放综合征等CAR-T细胞治疗的常见不良反应有明显区别。由于细胞放行检查记录缺失,无法排除细胞质量不达标准的可能,增加了细胞回输后不良事件原因的分析难度。
CDE生物制品临床部审评员高建超指出,良好的细胞质量控制是开展临床试验的基本前提。在开展注册临床试验时,必须在确保细胞产品符合相关质量控制要求的前提下开展研究,避免因细胞质量原因产生的安全性风险。
对于细胞治疗临床风险的控制,也体现在每一个环节中。高建超表示,免疫细胞产品回输后安全性风险暴露需要一定时间,患者之间如果没有设置足够的观察间隔,将存在非常大的安全性隐患。申请人在开展剂量探索试验时,必须根据细胞产品的作用特点设置足够的观察间隔,以降低受试者参与临床试验的安全性风险。
他以CAR-T举例说明:根据CAR-T细胞回输后细胞因子释放综合征、神经毒性等常见不良反应的发生规律,在早期剂量探索研究中,建议每一剂量水平的受试者入组间隔不少于2周,以观察患者回输后的安全性和耐受性情况。
另外,高建超强调了风险控制方案和受试者选择的重要性。在临床试验方案中,申请人应结合国内外临床共识和研究进展,针对可能出现的安全性风险的预防、识别、诊断、治疗及预后随访等制定全面合理、可操作性强的风险控制方案(RMP),并通过完善的研究者和医务人员培训确保临床试验参与者严格执行方案中的相关操作流程。
试验方案中的入排标准不仅仅是受试者参加临床试验的门槛,更是对受试者风险承受能力的综合评价。申请人和研究者应保证受试者充分了解参加细胞治疗临床试验的风险,不应夸大细胞治疗的临床获益、刻意淡化不良反应风险。
科研思路到药物开发的转变
以科研的思路进行药品的申报,也是多位CDE专家在工作中发现的共性问题。
在药学专业审评员看来,研究要尊重药品研发规律,不能急于求成,一些申报资料是从实验室研究转向药品研究,需要一定时间和数据积累,才能形成规范的、符合药物规模化研究与生产的资料,很多时候,这个过程还没有完成,申报者就急于报进来。
比如,在药学研究中出问题最多的生产用原材料部分,申报资料要求评价材料添加的必要性、合理性和安全性。有一些企业申报的资料中,血清添加直接按照2%或者5%的比例,没有自身的研究资料,这不是做药品的思路。
“做科研的时候可以采用别人的经验,但申报药品,在借鉴别人经验的基础上,还一定要自己开展产品的验证研究,验证具有风险的生产用原材料的添加量是一个最低的有效量才可以使用。现在很多资料中都是基于一种认知或他者的经验,缺少研究,这样是不合适的。”该名药学专业审评员说。
而对于非临床研究,所用样品需采用临床拟用样品,如果CMC发生变更,需进行变更前后的可比性研究,若变更前后CMC不具有可比性,可能需进行额外的非临床研究。
CDE药理毒理专业一位审评员还提醒申请人注意,细胞治疗产品的物质组成及作用机制与小分子药物、大分子生物药物不同,所以传统、标准的非临床评价方法可能不完全适用于细胞治疗产品,在非临床的研究中建议根据产品的特点进行相关非临床研究设计。
以CAR-T产品为例,其非临床研究应包括概念验证研究、药代研究与安全性研究等。概念验证研究应包括体外与体内研究,进行相关的机制研究及抗肿瘤活性研究,以评价产品的可行性和合理性。
非临床药代研究需评估产品在体内的增殖、存续时间和生物分布。非临床安全性评价应遵从GLP规范。安全性研究中,需根据产品特点及非临床药代特征设计合理的观察期,以支持临床试验方案,而非简单套用常规的急性毒性评价方法,还应关注CAR-T产品的脱靶毒性及基因修饰导致的插入突变的风险。
非注册临床研究数据
如何应用到药物申报
由于中国细胞治疗监管方面双轨制的特点,有些细胞治疗产品在向CDE递交注册临床申报之时,已经具有一些非注册的临床试验数据。如何在注册临床中应用这类数据,从而加快审批流程一直是企业最关注的问题之一,CDE接到过很多这方面的询问。
CDE生物制品临床部审评员黄云虹表示,这类临床试验结果如果要用于支持药品申报,需要关注生产工艺、质量控制、质量可比性的研究。因为如果想知道非注册临床试验所用产品与药物临床试验所用产品是否一样,需要对这些研究数据进行评价。
药学专业审评员也表示,开展非注册临床研究的产品准备IND时,它们之间的工艺和产品质量的情况要说明,如果发生变化一定要进行桥接研究。非临床动物研究和非注册临床试验研究的数据,只有在桥接研究的基础上,才能成为评价产品能否进入临床的基础。
关于非注册临床研究获得的人体数据对非临床研究的意义,药理毒理专业审评员认为,非注册临床研究需符合CDE的相关要求,经相关专业科学评估后,如可提示产品的有效性与安全性特征、临床风险可控,提交相关的体外有效性与安全性研究信息后,可接受开展临床试验,并在临床期间需进一步完善非临床研究(包括体内研究)。作为产品后续临床研究及上市申请的有效性与安全性支持性信息。如果非注册临床研究产品不符合相关要求,则需提供全面的非临床有效性与安全性研究资料。
黄云虹还强调:“由于对非注册临床数据获得的过程、方案、操作不了解,所以CDE希望申请人能提供尽量详尽的数据,来证明试验是合规的,保证数据的真实性、完整性和数据质量。我们见过申请人只把某些数据提交给我们,然后在一个国际大会上发布同一试验的数据,跟提交给我们的不完全相同,这是不可以接受的。”
这也是CDE多位专家在会上反复提及,申请人应与药监部门保持良好的沟通交流。除了充分利用沟通交流机制,非常关键的一点是申请人需要向CDE提交完整和真实的资料和数据,如果隐瞒核心的资料或者真正想用的工艺,沟通就是无效的。
来源:研发客 作者:程昊红

为你推荐
 资讯
资讯 CDE:治疗用重组蛋白药物首次申报临床试验药学资料撰写指导原则
本指导原则基于 ICH M4Q( R1)总体框架, 格式体例与之保持一致, 在其框架下结合治疗用重组蛋白药物的药学研究特点,细化了 IND 申报药学资料的撰写要求,旨在为该类药...
2026-04-24 12:44
 资讯
资讯 甘李药业GLP-1R博凡格鲁肽Ⅲ期临床试验完成首例受试者给药
4月23日,甘李药业发布公告,博凡格鲁肽(研发代号:GZR18)注射液正在中国开展的适应症成人肥胖患者的中度至重度阻塞性睡眠呼吸暂停(OSA)的Ⅲ期临床试验,于近日成功完成首例...
2026-04-24 10:25

 资讯
资讯 应世生物再冲港交所 IPO:手握国内唯一 III 期 FAK 抑制剂,专攻肿瘤耐药后市场
公司战略性聚焦黏着斑激酶(FAK)及整合素通路,这些靶点对肿瘤细胞的顽强生存能力至关重要。
2026-04-23 22:00
 资讯
资讯 华东医药2025年创新收入大增64.2%,2026Q1扣非净利创单季历史新高
2026年4月23日晚间,华东医药(000963 SZ)发布2025年年度报告及2026年第一季度业绩。
2026-04-23 21:45
 资讯
资讯 默克与谷歌云达成一项10亿美元的AI合作
此次合作,默克期望将自身在药物研发、科学管理、市场运营等方面能力与人工智能、云平台相结合,帮助默克全球约7 5万名员工提升生产力。
2026-04-23 13:00
 资讯
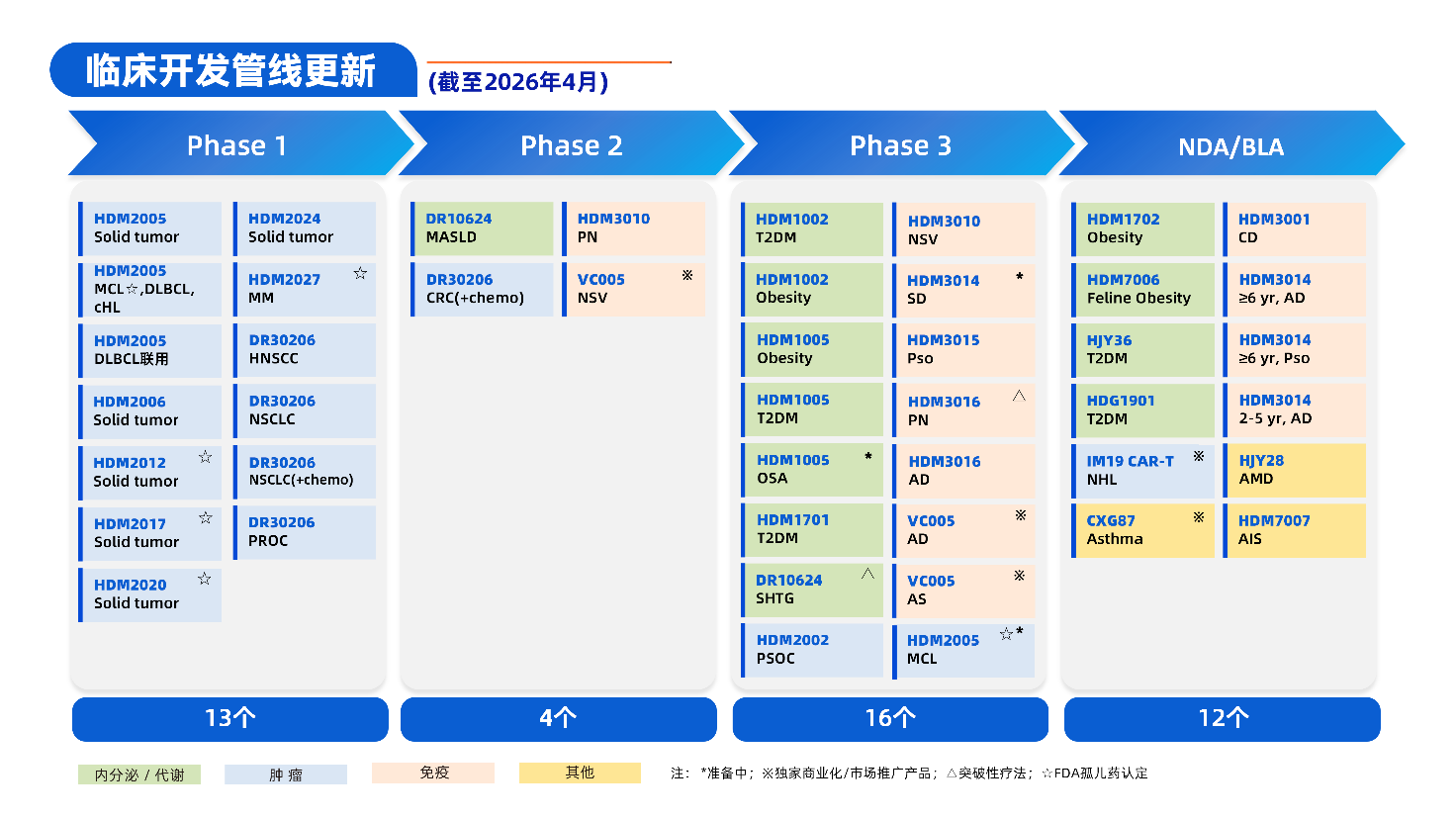
资讯 神经介入与神经外科植入材料等7类医用耗材分类与代码及医保通用名
近日,国家医保局发布《神经介入与神经外科植入材料等7类医用耗材分类与代码及医保通用名》公开征求意见的公告。
2026-04-23 11:29
 资讯
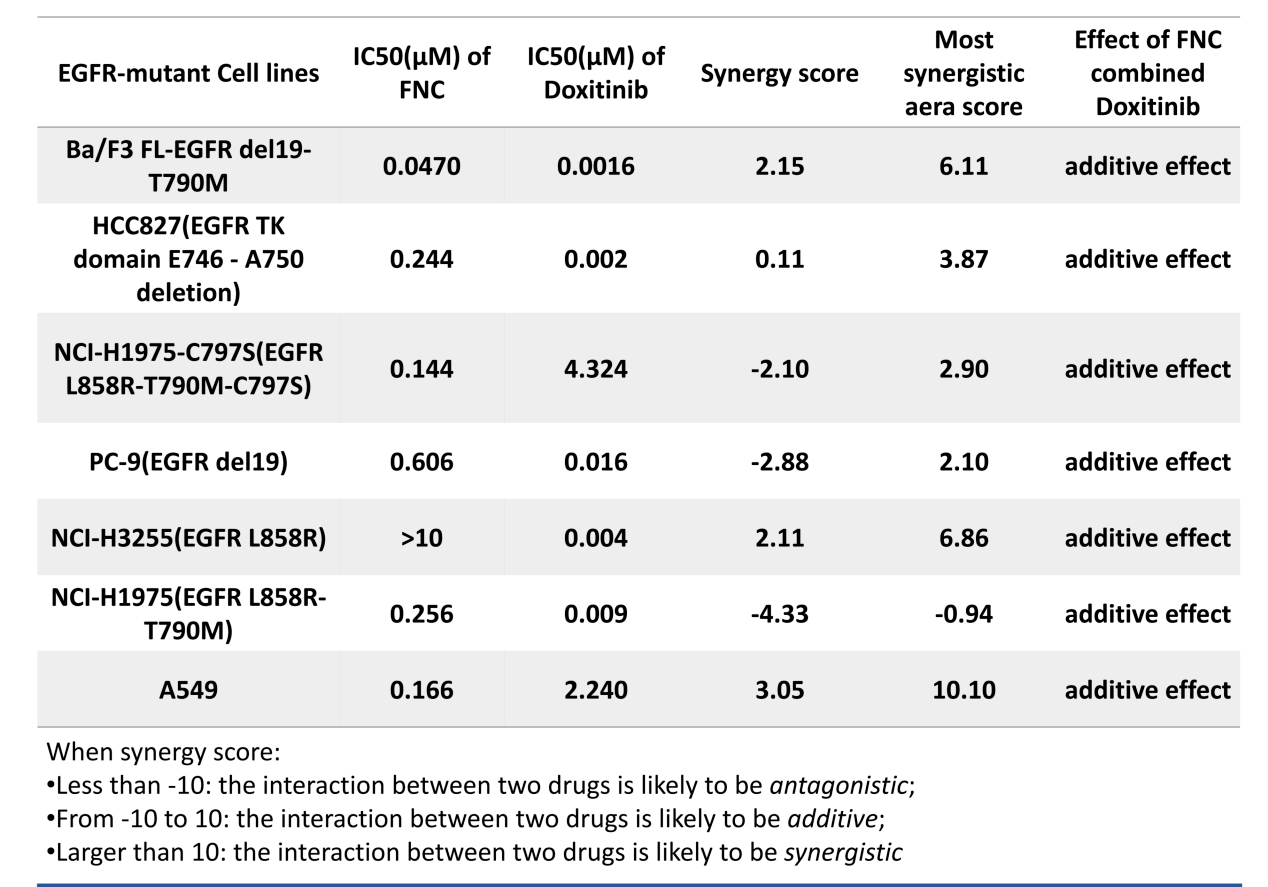
资讯 真实生物阿兹夫定与哆希替尼联合疗法最新消息
近日,真实生物在美国癌症研究协会(AACR 2026)年会以壁报形式展示了阿兹夫定与哆希替尼联合疗法的临床前研究成果,标题为标题:Azvudine Combined with Doxitinib,a Potential Therapy for EGFRm+ NSCLC。
2026-04-23 11:07

 资讯
资讯 华东医药独家商业化产品CXG87上市申请获受理,呼吸治疗赛道布局深化
华东医药股份有限公司(以下简称“华东医药”或“公司”)全资子公司华东医药(杭州)有限公司独家商业化产品CXG87(布地奈德福莫特罗吸入粉雾剂(IV)胶囊型)用于治疗哮喘的药...
2026-04-22 17:53
 资讯
资讯 指南更新,巨星发声:医疗创新与公众教育双轮驱动OSA诊疗革新
日前,中国《成人阻塞性睡眠呼吸暂停诊治指南(2025)》发布,首次系统性引入药物治疗——GIP GLP-1双受体激动剂替尔泊肽获最高级别推荐,用于合并肥胖症的中至重度阻塞性睡眠...
2026-04-22 10:04
 资讯
资讯 江苏2025年癌症发病情况
近日,江苏省发布2025年癌症发病情况数据。据2025年最新肿瘤监测数据显示,江苏省癌症发病率为392 84 10万,较2024年(380 50 10万)增长3 24%。同期江苏全省癌症死亡率为2...
2026-04-21 16:39
 资讯
资讯 中国生物制药发布了其PD-1/VEGF双抗的初步临床试验数据
4月19日,中国生物制药发布了其PD-1 VEGF双抗(药物研发代号为MK-2010 LM-299)的初步临床试验数据。
2026-04-21 13:25
 资讯
资讯 天境生物将菲泽妥单抗大中华区权益转售给渤健
4月20日,天境生物(TJ Biopharma)与渤健(Biogen,纳斯达克代码:BIIB)共同宣布,双方已达成一项最终协议,渤健将从天境生物获得菲泽妥单抗的大中华区(包括中国大陆、香港...
2026-04-21 11:00
 资讯
资讯 云顶新耀发布EVM16个性化肿瘤治疗性疫苗首次人体临床试验数据
4月20日,云顶新耀在2026年美国癌症研究协会(AACR)年会上公布了其自主研发的mRNA个性化肿瘤治疗性疫苗EVM16单药治疗及联合PD-1抑制剂(替雷利珠单抗 百济神州)治疗晚期实体...
2026-04-21 09:15
 资讯
资讯 剂泰科技通过港交所聆讯,携全球首个 AI 纳米递送平台冲刺 "AI 药物递送第一股"
剂泰科技成立于 2020 年,由美国工程院院士陈红敏博士以及 MIT 科学家赖才达博士、王文首博士联合创立,是一家专注于 AI 驱动纳米材料创新的高科技企业。
2026-04-20 17:25
 资讯
资讯 飞眸医疗完成近亿元天使轮融资,加速打造国际领先个性化全飞秒屈光系统平台
近日,深圳市飞眸医疗器械技术有限公司(以下简称 "飞眸医疗")正式宣布完成近亿元天使轮融资,本轮融资由元生创投领投,清科控股、清科大
2026-04-20 17:18
 资讯
资讯 倍乐锐(注射用玛贝兰妥单抗)获批用于治疗二线及以上多发性骨髓瘤成年患者
与达雷妥尤单抗的三联疗法相比,倍乐锐®联合治疗方案可降低死亡风险42%,并延长中位无进展生存期近三倍。
2026-04-20 16:58