在过去的十几年中,生物技术和制药企业给全球市场带来了一种新的治疗方法–生物类似药(从欧盟到印度到韩国到美国),为一些最昂贵的药品节约了成本。即使在2018年,距各企业开始寻求开发生物类似药已经过去了几十年,生物类似药仍然只是刚刚开始被广泛采用。
什么是生物类似药?
生物类似药,也被称为生物仿制药,是与已批准的生物原研药相似的一种生物药(包括疫苗、血液及血液成分、体细胞、基因治疗、组织和重组治疗性蛋白等)。
FDA指出,生物制剂和生物类似药都是从各种自然资源中分离出来的——人、动物或微生物——也可以由糖、蛋白质、核酸或这些物质的复杂组合组成,或者它们甚至可能是生物体,如细胞和组织。
在美国和欧盟这两个目前最大的生物类似药市场,生物类似药必须与已批准的原研药在质量、安全性和有效性方面没有临床意义的差异(在美国,这种已授权的产品被称为参比制剂)。
同样的,在这两个市场,企业需要开展研究,证明生物类似药与参比制剂类似,在质量、安全性和有效性方面不存在任何有意义的差异。
不过,EMA指出,原研药为生物类似药的使用、生产提供了数据,意味着批准生物类似药所需的安全性和有效性数据量通常比批准原研药所需的要少。
生物类似药与化学仿制药的不同之处?
仿制药 (必须是相同的参考产品)和生物类似药之间的争论日益激烈,一些媒体称生物仿制药为“山寨”,或“冒牌货”。
要想了解生物类似药和化学仿制药的差异,首先需要弄清楚原研生物药和化学药的差别。小分子化学药通常是化学合成的,而大分子生物药则通常是生物合成的。源头的不同就直接造成两者在结构、成分、生产方法和设备、知识产权、配方、保存方法、剂量、监管方式以及销售方式均有不同。
与合成的小分子化学药相比,生物药在分子大小上要大一百至上千倍。比如抗体药分子量高达15万道尔顿,而化学药通常不到1000道尔顿。有报道将小分子化学药的大小比作一辆自行车,而生物药的个头则相当于一架飞机,其实两者的区别不仅仅是分子大小的差别。更重要的是,生物药的分子结构要远比化学药复杂。
由于生物药具有更大的分子量和复杂的结构,生物药的表征面临很大挑战。但由于上述特点,即使目前全世界最先进的仪器设备全用上,也不可能将生物药的结构等特性完全表征清楚。这些特点也注定生物类似药不可能完全和原研药一模一样,即使是同一家公司生产的同一种生物药,不同批次也会有差异。即使是同一批次,在储存、流通的过程中,生物药(尤其是蛋白类药物)的结构和活性也不可避免地会有所变化。
对于生物类似药生产商而言,由于知识产权保护等多种原因,原研药公司所采用的生产工艺甚至是所采用的细胞系都会不清楚,这就更导致生物类似药与原研药不会一样。另外,对于生物药而言,其生产及流通过程更加复杂,要求也更高,有许多步骤,细胞培养的条件(温度和营养)、产品的加工、纯化、储存和包装等各个环节都会影响产品的生产,整个过程中的微小差别都可能会对最终产品的质量、纯度、生物特性以及临床效果产生较大影响。正由于上述种种原因,虽然化学仿制药的英文是generic drug,但是生物类似药并非是biogeneric,而是biosimilar,因为生物类似药只可能与原研药“相似”(similar),绝不可能一样。
总的来说,生物药的生产对于其生产条件的要求远比化学药苛刻,当然生产成本也更高,而且生物药的临床前和临床阶段的研发成本也更高。
不过,据IMS,生物类似药对一些欧盟国家的重大影响正逐步显现(见下图)

对美国而言,潜在的成本节约仍有待确定,不过目前的估计将不得不考虑到后来在这个解释中所提到的一些问题。
IQVIA还提供了第一个生物类似药在未来10年将会出现的地方:

CVS在一份报告(下图)中列出了许多其他与成本相关的因素。

随着生物类似药通道的不断扩大,更多的竞争可能意味着更低成本的治疗(尽管许多生物类似药已经被批准或提交审批,因为这张图表已经发布了——更多的是关于批准的)。

有哪些生物类似药在哪儿获批了?
在欧盟,生物仿制药的批准相对较多,(欧盟只能批准参比原研药已经上市10年的生物类似药,美国则是需要原研药已上市12年;但在美国,一个生物类似药可以在参比原研药获批后4年申请FDA备案)。下表是2006年12月至2018年1月获批的超过20种生物类似药:






与他们的参考产品相比,Remicade生物仿制药现在占欧盟市场份额的53%,伯恩斯坦(Bernstein)的生物技术分析师罗尼·格尔(Ronny Gal)表示,虽然Enbrel生物仿制药的市场份额为29%,rituximab (rituximab)的仿生物仿制药(rituximab)仍未在美国获得批准,但已经获得了13%的市场份额。
然而,在美国,FDA已经授权使用9种生物仿制药(实际上只有3种已经上市):Sandoz's Zarxio (filgrastim-sndz),与Amgen的Neupogen类似,Enbrel与Erelzi (etanercept-szzs)的竞争,后者尚未进入美国市场,并在2029年获得专利保护; Avastin与Mvasi (bevacizumab-awwb),在美国直到2019年初才会进入市场;对于Humira, Amjevita(adalimumab-atto)和Cyltezo (adalimumab-adbm)和Herceptin与Ogivri (trastuzumab-dkst),在2017年12月获得批准,但发行日期仍然未知。辉瑞的Ixifi (infliximab-qbtx)也于2017年12月获得批准,不过该公司表示不会推出该产品。
Remicade也有两种生物相似药的竞争对手,FDA批准的——三星Bioepis公司的Renflexis (infliximab-adba)和Celltrion和辉瑞公司的Inflectra (infliximab-dyyb),这两家公司都已经上市,但它的市场份额并不大,仅占市场份额的5%。另一个唯一发行的生物类似药是Zarxio (filgrastim-sndz)。
FDA可能在未来几年批准更多的生物类似药, 在美国,生物类似药的概念很棘手,因为一些被批准的生物制品,如胰岛素,有时被认为是生物类似药,尽管FDA曾表示,在某些情况下,与礼来的Basalgar一样, 它们并不是生物类似药,因为它们是通过505(b)2的应用程序获得批准的,这在一定程度上依赖于赛诺菲的Lantus的应用。
截止到2018年1月,60个生物类似药被纳入FDA的生物类似药开发项目,FDA已收到会议要求,讨论27种不同参考生物制剂的生物类似药的开发。
在审批数量方面,印度实际上是走在欧盟前面,自2000年来,已批准超过60个生物类似药。印度的CentralDrugs Standard Control Organization于2012年发布相关指南文件,2016年3月更新。
日本的卫生,劳动和福利部(MHLW)在2009至2016年间批准了9个生物类似药(包括胰岛素),并在2009年发布生物类似药行业指南。
韩国的食品药品安全部 (MFDS)在2012年至2015年间批准了6个生物类似药,也根据欧盟、日本和WHO的生物类似药行业指南,在2009年发布了指南文件。
批准途径
在美国,生物制品许可申请(BLA)执行21 CFR600–680规定(表356h细化了BLA的要求)。
根据2009年生物制品价格竞争和创新法案(BPCIA),2010年3月,作为《病人保护与平价医疗法案》(即奥巴马医改)的一部分,一种生物类似药于FDA批准的生物制剂(参考产品)的生物制剂可以使用《公共卫生服务法案》(PHS Act)第351(k)条的缩写批准途径。
FDA的生物类似药研究负责人LeahChristl在2016年6月解释说: “数据包在发起者和生物类似药的营销应用程序之间存在差异,但是在351(a)和351(k)项下的生物类似药的批准标准是相同的,这两者必须证明它们是安全的、纯粹的,并且能有效地满足被批准的使用条件。”
此外,她说:
1. 建议生物类似药的申办方不需要独立地确定其产品的安全性和有效性;它通过对参考产品的生物相似性的演示来建立这一点。
2. 建议生物制剂的申办方证明其与参考产品相似,可以依赖于FDA以前的安全、纯度和效力的发现(即:,安全性和有效性)参考产品。“依靠某些现有的科学知识来支持安全性、纯度和效力(即:生物类似药产品的安全性和有效性允许一个可能更短、成本更低的药物开发项目。这是一个简短批准途径。
3. 这个简短途径并不意味着低标准
4. 数据包不同于发起者(351(a)BLA)和生物类似药 (351(k) BLA)营销应用程序,但是,原研药和生物类似药的批准标准是相同的,这两者必须证明它们是安全的、纯净的,而且对批准的使用条件是有效的。
5. 生物类似药的批准所需的数据包相当广泛;这是被缩写的批准途径。
2016年5月,赛诺菲(Sanofi)、迈兰(Mylan)、诺和诺德(Novo Nordisk)和行业组织PhRMA和生物仿制药理事会(thebiosimilar Council)呼吁FDA修改其对”被认为是许可证“条款的解释,因为一些人表示,目前的指导草案可能会在很长一段时间内停止生物类似药的开发。
除了351(k)通道,联邦食品、药物和化妆品法案,根据2012年生物类似药的用户费用法案(BsUFA)修订,FDA可以从2012年10月到2017年9月评估并收取生物类似药的费用。用户收费计划于2017年8月重新获得授权,并将在2022年通过额外收费。
在欧盟这一边,早在2003年就已经有生物类似药的申请通道了。
”评价的主要部分是将生物类似药与参考药物作对比,显示无显着性差异,“EMA说。”有关的监管机构在他们的评价研究中,采用严格的标准,比较两种药物的质量、安全性和有效性。质量的研究包括其活性物质的结构和生物活性的综合比较,安全性和有效性的研究要表明他们的利益和风险,包括免疫反应的风险没有显着差异。“
EMA说,和美国一样,因为参考药物在欧盟已经被授权了10年(美国12年) 它的临床效益已经很好地建立起来,一些与参考药物进行的研究可能不需要被复制。
命名
在欧盟,生物类似药使用独特的专有名称,与其参考药品相同的通用名(也被称为国际非专利名称或INN),就像化学仿制药那样。例如,Biogeand Samsung的Benepali(2016年1月在欧盟批准)是安进公司的Enbrel的生物类似药,都使用依那西普(etanercept)作为非专有名称。
然而在美国,生物类似药的命名却引起了争议,主要是因为一条要求(2015年8月发布的命名指南)在生物类似药的非专利名末尾加上四个字母的后缀(因此- dyyb和-sndz出现在两个已经批准的生物类似药的末尾)。不过由于这一命名提议遭到各方异议,因此目前暂时建议在生物类似药的非专利名末尾加上1至3个字母的后缀以将其和原研药区别开来。
行业组织对该指南进行了评论,包括生物仿制论坛,PhRMA和BIO,支持FDA的提议,但要求该机构使用”有意义的“和”可区分的“后缀与许可证持有人的名字联系在一起,就像第一个被美国批准的生物类似药,被称为Zarxio (filgrastim-sndz)。
WHO则草拟了一份提议,提议用”生物品资质(biological qualifier)“将生物类似药与原研药区别开
标签
在2016年3月,FDA公布了生物类似药标签的指导草案,这将严重依赖于他们的参考产品标签,尽管生物制品行业可能会高兴的是,标签必须对生物相似和参考产品做出澄清。
FDA说,与仿制药一样,生物类似药的标记应该包括对临床数据的描述,这些数据支持参考产品的安全性和有效性。
与普通药物不同的是,生物类似药的参考产品没有得到相同的适应症。FDA注释,”可能有必要在生物类似药产品标签中包含相关信息,而生物相似药产品申请人并不是为了确保安全使用而寻求许可。“(例如,当参考产品标签上的安全信息与产品的使用有关,并不是特定于某一特定的批准指示,或当仅针对生物类似药产品的指示信息时,不能轻易提取)。
该指南称,该标签应该以一种并不意味着生物类似药产品被批准用于参考产品指示或未被批准用于生物类似药产品的方式来书写。
FDA补充说:”参考产品的总体风险收益情况与生物类似药产品相关,即使在产品标签上可能没有出现与生物相似产品相关的严重不良反应或其他风险。“
FDA还提供了一个虚构的参考产品JUNEXANT (replicamab-hjxf)和一个虚构的生物类似药产品NEXSYMEO (replicamab-cznm)。

州立法
尽管BPCIA建立了生物类似药能够进入美国市场的途径,但国家(深受生物制药行业的说客影响)已经着手将生物类似药代入他们自己的手中。
24个州(最近的一次是在2016年7月的宾夕法尼亚州)和波多黎各现在已经制定了法律,制定了一系列关于生物类似药如何分配的法规,甚至在生物类似药在美国市场大规模上市之前,FDA已经解释了什么是可交换性。
法律一般要求药剂师只能在FDA认定生物类似药可互换的情况下,才可替代其参考产品。如果开药者并不禁止这种替换,如果替换的药房通知病人,如果药店保留生物类似药分发的记录,并在五个工作日内发放生物类似药,药剂师记录提供给病人的是什么具体的产品,包括产品名称和制造商名称。

古德温(Goodwin)波士顿办事处的诉讼部门主管伊莱恩?布瑞斯(Elaine Blais)在2016年1月表示,生物制药公司一直”疯狂地“在游说州立法机构,以使自动替换变得困难。
她说:“我无法想象这些钱会花在这上面。”
来源:HPC药闻药事
作者:RAPS HPC
来源:HPC药闻药事 作者:RAPS HPC

为你推荐
 资讯
资讯 章科:坚持为人民出政绩 推动医疗保障事业高质量发展
医疗保障制度依法保障每一位参保群众的医保权益,有力提高全社会医疗服务的可及性,有助于增进民生福祉、缩小贫富差距,促进社会公平和共同富裕。
2026-08-05 10:48
 资讯
资讯 康宁杰瑞与PathosAI达成22亿美元ADC授权协议,1.25亿美元首付款
8月4日,康宁杰瑞生物制药宣布其全资子公司江苏康宁杰瑞生物制药有限公司与美国临床阶段生物技术公司Pathos AI就TROP2 HER3双特异性抗体偶联药物JSKN016达成独家授权许可协议。
2026-08-04 22:14
 资讯
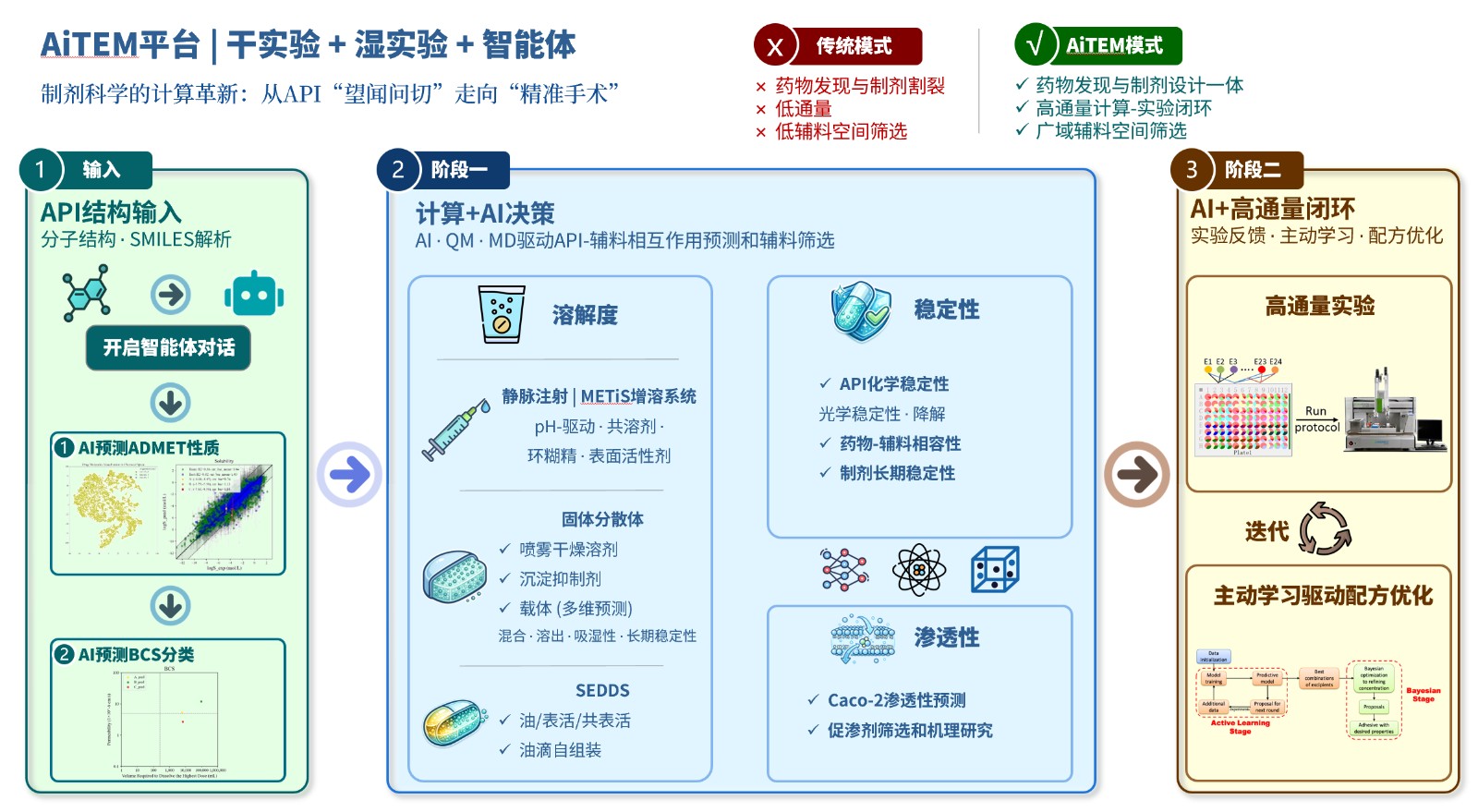
资讯 剂泰科技与恒瑞医药达成AI制剂合作
8月3日,剂泰科技发布自愿公告,宣布自主研发的AiTEM人工智能制剂创新平台将在恒瑞医药进行本地化部署,加速药物制剂配方开发效率。
2026-08-04 13:01

 资讯
资讯 东阳光药甘精胰岛素首批115.2万支发货美国
近日,东阳光药发布公告,首批甘精胰岛素注射液(商品名:Langlara)已于7月27日自湖北宜都生产基地分批启运美国,预计8月中下旬完成全部发货。首批计划发货115 2万支。
2026-08-04 11:37
 资讯
资讯 维利信LBL-024用于转移性结直肠癌获批IND
8月3日,维立志博发布公告,公司自主研发的PD-L1 4-1BB双特异性抗体维利信(LBL-024)联合用药治疗转移性结直肠癌(mCRC)的开放、多中心Ib II期临床试验申请已获得国家药品监...
2026-08-04 10:22
 资讯
资讯 药明康德半年业绩超预期,大幅上调全年业绩指引
预计2026年公司整体收入由人民币513-530亿元上调至人民币585-605亿元,上调幅度约14%,持续经营业务收入同比增速由18%-22%上调至35%-39%。药明康德计划向全体股东每10股派发现金红利5 1元(含税)。
2026-08-03 20:46



 资讯
资讯 国家医保局召开全国深化医保基金管理突出问题专项整治工作中期推进会
7月31日,国家医保局会同最高人民法院、最高人民检察院、公安部、财政部、国家卫生健康委、税务总局、市场监管总局、国家中医药局、国家药监局,10部门联合召开全国深化医保基金...
2026-08-03 10:40
 资讯
资讯 第12批国采,哪家企业中选品种最多?
本次集采65个品种全部采购成功,全国共4 5万家医药机构报量,327家企业的521个产品获得拟中选资格,覆盖抗感染、抗肿瘤、抗血栓、降血糖、降血压、降血脂、风湿免疫、消炎镇痛...
2026-08-02 19:58
 资讯
资讯 雷海潮:推动健康中国建设取得决定性进展
健康中国建设是一项系统工程,涉及面广,必须充分发挥党总揽全局、协调各方的领导核心作用。
文/雷海潮 国家卫生健康委员会党组书记、主任 2026-08-01 22:28
 资讯
资讯 华恒生物实控人被批准逮捕
8月1日,华恒生物发布《关于重大事项进展的公告》,公司于2026年7月31日收到公司实际控制人郭恒华家属通知,郭恒华因涉嫌非法吸收公众存款罪被检察机关批准逮捕。
2026-08-01 17:47
 资讯
资讯 国务院常务会议:研究健康优先发展战略实施有关工作
要牢固树立健康优先导向,深化医药卫生体制改革,健全公共卫生、医疗服务和医疗保障体系,推动医药科技创新,加快形成有利于健康的生产生活方式。
2026-08-01 10:25
 资讯
资讯 总计最高4.235亿欧元付款,英派药业与Pharmanovia达成授权合作
7月30日,南京英派药业公告称,公司已与Pharmanovia订立独家合作,授予其在66个国家制造、开发及商业化塞纳帕利的独家权利。公司有望获总计最高达4 235亿欧元付款,包括首付款...
2026-07-31 16:12