
1、概 述
阿达木单抗(Adalimumab)系在中国仓鼠卵巢细胞中表达的重组全人源化肿瘤坏死因子α(Tumor Necrosis Factor, TNFα)单克隆抗体注射液,由美国雅培公司研发上市,商品名为:修美乐(Humira)。阿达木单抗在美国和欧盟已获批多个适应症 [1,2],2010年首次获准进口中国。目前在中国批准用于:①对改善病情抗风湿药(DMARDs),包括甲氨蝶呤疗效不佳的成年中重度活动性类风湿关节炎患者(RA);②常规治疗效果不佳的成年重度活动性强直性脊柱炎患者(AS);和③需要进行系统治疗的成年中重度慢性斑块状银屑病患者(Ps)。[3] (见表1)。
阿达木单抗注射液原研产品美国专利已于2016年到期,欧洲专利2018年即将到期[4],国内外制药企业已纷纷加入其生物类似药的研发。Amgen研发的ABP501(Amgevita/ Amjevita)、BI研发的BI695501(Cyltezo)、Samsung Bioepis研发的SB5(Imraldi)和Sondoz研发的GP2017(Hyrimoz?)均已作为阿达木单抗的生物类似药在欧盟获批上市
[5-8],其中的ABP501、 BI695501和GP2017在美国也已按生物类似药获准上市[9-11]。
本文在原国家食品药品监督管理总局已发布的《生物类似药研发与评价技术指导原则(试行)》[12](以下简称“指导原则”)基础上,结合阿达木单抗的特点,重点探讨当前普遍关注的临床研究策略和临床试验设计问题,以期为国内阿达木单抗生物类似药的临床研发提供参考。
2、阿达木单抗生物类似药临床研究策略
根据《指导原则》,生物类似药研发总体思路是通过系统的比对试验证明候选药与原研药的相似性为基础,支持其安全性、有效性和质量可控等方面与原研药的相似性。依据逐步递进的原则,分阶段进行药学、非临床、临床比对研究。进行阿达木单抗生物类似药临床研发的首要前提是已通过前期药学和非临床比对试验证明候选药与原研药相似,在此基础上方可按照生物类似药的路径开展药代动力学比对试验和临床安全有效性比对试验。
原则上,药代动力学比对试验需要进行1项健康受试者单次给药药代动力学生物等效性研究,验证候选药与原研药PK特征的相似性。临床比对研究需选择国内已经获批适应症人群,与原研药进行1项“头对头”比较的临床等效性研究以支持其注册上市。按此临床研发思路完成单个适应症的临床比对研究可寻求外推其它相同作用机制适应症,同时应考虑其与原研药的整体相似性。
3、阿达木单抗临床研究设计要点
生物类似药临床比对研究设计应当以证明候选药与原研药的相似性为目的,进行科学合理的研究设计。临床研究中应采用与国内进口相同来源的原研药。如果选择其他来源的原研产品,应提供与国内进口阿达木单抗原研药的可比性证据。
(一)健康受试者药代动力学比对研究
试验设计:阿达木单抗半衰期较长(约2周)[3],且具有潜在免疫原性等特征,建议参照一般生物等效性研究的设计,采用单次给药的随机、双盲、平行对照的试验设计评价其PK特征的生物等效性。
研究人群:健康志愿者是较为理想的均质性受试人群,能更好的反映出候选药与原研药之间PK特征的一致性,仅选择健康成年男性志愿者是可行的。建议尽量控制受试者年龄和BMI指数在相对较窄的范围内,以期受试者人群相对均一。
剂量及给药途径:选择的给药剂量应能敏感地分辨候选药和原研药PK特征差异。根据国外阿达木单抗生物类似药PK比对研究经验,推荐与国外采用相同研究剂量,即:40mg/0.8ml,皮下注射。建议选择统一的注射部位。
终点指标与界值:PK比对研究主要终点指标的选择是等效性评价的关键。根据口服固体制剂的相关指导原则[13],AUC0-∞和Cmax是判断生物等效性的主要参数。但是在生物类似药的生物等效性评价中,选择AUC0-t还是AUC0-∞作为终点尚未形成一致意见。目前的观点认为AUC0-t是通过实际测量值计算获得的,考虑到生物类似药药代动力学的特性和实际研究过程中取血点设置的相关性,推荐AUC0-t和Cmax作为主要研究终点指标,AUC0-∞、tmax、Vd和t1/2作为次要研究终点重点进行比较分析,等效性界值建议设定为80%-125%。
样本量:通常90%置信区间可接受的等效性判断界值为80%-125%,估算样本量时把握度可以取80%。还应结合原研药既往信息及药代参数变异情况综合考虑。
(二)临床有效性比对研究
临床比对研究适应症人群的选择应基于以下基本考虑:
(1)患者的免疫能力,伴随用药(例如甾体类药物、甲氨蝶呤等),免疫的敏感性和患者人群的均质性;
(2)有效性终点的敏感性,一般会优先选择“客观评估标准”,“持续性,可评价的终点”;
(3)伴随治疗的一致性和稳定性;
(4)被选择的适应症历史临床数据的充分性和变异性。目前,国外进行阿达木单抗生物类似药临床比对研究时多选择RA、Ps患者作为研究人群。
试验设计:临床比对研究的目的是证明与原研药临床疗效的相似,推荐采用等效性设计,以进口原研药为对照,进行随机、双盲、平行对照试验。
1. 选择类风湿关节炎患者人群(RA)
诊断标准:目前, ACR1987年修订的分类诊断标准和2010年ACR/EULAR分类诊断标准在临床上均有应用。考虑到2010年诊断标准中包括C反应蛋白和血沉等客观指标,且国外阿达木单抗生物类似药临床研究中也多采用2010年ACR/EULAR分类诊断标准,具有较好的灵敏度和特异度[14],故推荐使用2010年ACR/EULAR分类诊断标准。
疾病活动度:
(1)国外已上市阿达木生物类似药临床比对研究时,对患者疾病活动度的要求一般为:≥6个肿胀关节(基于66个关节计数)和≥6个压痛关节(基于68个关节计数);血沉(ESR)≥28mm/hr或C反应蛋白(CRP)>10mg/L。
(2)考虑到国内临床诊疗实践变化和DAS28评分已广泛用于疾病活动度评估,疾病活动度评估满足DAS28≥3.2且CRP>10mg/L的RA患者也可以作为阿达木生物类似药临床比对研究受试者。为保证研究人群的均质性,在1项研究中只能采用上述2种疾病活动度要求的1种。
背景治疗:RA患者应满足连续接受甲氨蝶呤( MTX)治疗≥3个月且使用稳定剂量MTX(≥10mg/周)持续治疗不少于4周。
给药方案/剂量:建议按原研药进口说明书中批准的给药方案和剂量给药,即:40mg/0.8ml/次,每两周一次皮下注射。治疗期间继续使用稳定剂量的MTX。
主要终点指标:主要终点的选择应基于能证明候选药与原研药临床相似且能敏感甄别出两者临床疗效差异。国外已批准的阿达木单抗生物类似药在RA患者中进行的临床比对研究一般选择治疗趋于稳定的第24周ACR20应答率作为主要终点指标且用来确定相应等效性界值的数据来自于第24周ACR20应答率指标[15,16]。因此,推荐第24周的ACR20应答率作为主要终点指标。如采用第12周ACR20应答率作为主要终点指标,应充分考虑该时间点ACR20应答率的变异度。
等效性界值:目前国际上计算设定界值时对使用候选药组与原研药组研究终点的差值(Risk Difference,RD)或者比值(Risk Ratio,RR)仍存在不同意见。国内外制药企业在进行RA患者人群的临床疗效比对研究设计时采用了不同的界值标准进行样本量估算。在阿达木单抗生物类似药的审评中,美国FDA和欧盟EMA接受基于RD的95%置信区间等效性界值,分别为±12%、±15%[17,18]。进一步分析全球临床研究数据和既往已获得的中国数据,建议RA适应症临床比对研究的等效性界值按RD的95%置信区间设定为±15%。
2. 选择强直性脊柱炎患者人群(AS)
诊断标准:近年来AS诊断标准多采用1984年修订的纽约标准。2009年ASAS专家组公布了新的中轴型脊柱关节炎的分类标准。考虑到1984年修订的纽约标准仍然是目前常用的诊断标准,且也是当前AS创新药临床试验中采用最多的诊断标准,故建议采用1984年修订的纽约标准。
疾病活动度:根据阿达木单抗国外关键性研究和进口注册研究[17,18],满足以下三项中的至少两项:BASDAI≥4cm、整体背痛(VAS)≥4cm和晨僵≥1小时。
背景治疗:对至少1种非甾体抗炎药(NSAID)疗效不佳或不耐受。 “疗效不佳”一般是指接受非甾体抗炎药连续2~4周规范治疗后。
给药方案/剂量:建议按原研药进口说明书中批准的给药方案和剂量给药,即:40mg/0.8ml/次,每两周一次皮下注射。
主要终点指标:阿达木单抗国外主要临床研究和进口注册研究的主要有效性终点指标均为第12周ASAS20的应答率[17,18],且确定的等效性界值数据也是以第12周ASAS20的应答率为主要疗效指标。因此推荐采用第12周ASAS20的应答率为主要终点指标,并建议将第24周ASAS20的应答率作为次要终点指标。综合免疫原性考察需要和国内临床研发实践,可以接受以第24周ASAS20应答率作为主要终点指标,但同时应将第12周ASAS20应答率作为次要终点指标考察。
等效性界值:经汇总阿达木单抗AS适应症人群国外关键性研究M03-607和进口注册临床研究M11-991临床疗效数据[17,18],试验组与安慰剂组第12周ASAS20应答率差值为37.3%(95%CI:29.9%,44.6%)。参照通常将参照药治疗效应差异下限的50%作为等效界值的确定原则,建议将ASAS20应答率的等效性界值按RD的95%置信区间设定为±15%。
(三)其他需要重点关注的问题
1. 安全性和免疫原性研究
免疫原性研究应贯穿在生物大分子药物整个研发过程中。免疫原性主要通过检测抗药抗体(anti-drugs antibodies, ADA)和中和抗体(Nab)的发生率来评价。
免疫原性试验结果与检测方法的敏感性,特异性及药物耐受性高度相关,并且可能受以下几种因素的影响:血样的处理、取样的时间、合并用药以及合并的疾病等。通常,临床免疫原性考察研究(包括ADA和Nab)与临床有效性比对研究在同一项临床试验中进行。推荐所有受试者均应进行免疫原性的考察,采样时间点设置应至少包括首次给药前,第4周或/和第12周,及末次给药后一个月,进而证实候选药在抗体阳性率、抗体滴度、抗体出现时间和中和抗体发生率等方面不高于原研药。同时,所涉及研究应证明生物类似药与原研药在免疫原性方面应不具有临床意义的差别。
安全性考察在药代和有效性比对试验研究中均应进行考察,对不良反应发生的类型、严重性和频率等进行比较,尤其是特定的重点关注的不良反应。
2. 患者药代动力学研究
通常,在进行患者临床比对研究时应同步开展多次给药PK研究,进而评估候选药与原研药在患者中的PK相似性趋势。PK采样点设置以能够较清晰地反映两者整体PK特征为原则。考虑到皮下注射给药的吸收过程,推荐患者多次给药的药代动力学研究在吸收到达稳态时进行采样,通过描述性统计,比较候选药和原研药之间药物暴露量(AUC0-tau、Ctrough,ss、Cmax)的相似性。
(四)适应证外推
适应证外推(extrapolation)是指在生物类似药研发中批准一个没有与原研药进行直接临床比对研究的适应症[19]。如果在原研药已批准适应症某一个人群中完成了生物类似药的系统比对研究,那么候选药就有可能基于已有的数据和信息寻求原研药已批准其他相同作用机制适应症的获批。适应证外推的前提是生物类似药与原研药的生物相似性已经被证实。适应证外推主要基于生物类似药比对研究所有可获得的数据和信息、原研药其他批准适应症临床研究在安全性和疗效方面的重要发现和对原研药每个适应症作用机制科学认知的综合考虑[19]。申报单位必须提供充分的科学证据以支持适应证外推的申请。
生物类似药在风湿疾病领域适应症的外推需要对所有比对研究数据进行评估后进行科学决策,不同品种和不同监管机构要求可能不同[20]。通常,不同适应症发病机制的差异,原研药在已批准不同适应症中的作用机制、PK、PD、疗效、安全性及免疫原性都需要给予关注。
4、小结
阿达木单抗生物类似药临床相似性研究应遵循生物类似药临床相似性评价的一般原则,即应当在有合理科学依据的前提下尽可能的简化,以能证实候选药与原研药相似性为目标,同时兼顾该品种的特性,进行有针对性的临床比对研究设计。鼓励研发企业与管理部门进行沟通,探索更加简便高效的研究设计方法。
本文主要观点已经相关领域专家,国内阿达木单抗生物类似药研发企业代表会议讨论并上网进一步征求意见,代表相关各方当前的共识。
[参考文献]
[1] U.S. Food and Drug Administration. Humira label(update 2017/12)
[2]European Medicines Agency. Humira information((updated on 15/12/2017)
[3]国家食品药品监督管理总局. 阿达木单抗注射液说明书. 2017.
[4] Sanjeev K. Gupta, Pankai S. Chaudhari, Rajalaxmi Nath. Opportunities and Challenges in Biosimilar Development [EB/OL]. (2017-05-18)[2018-02-09].
http://www.bioprocessintl.com/manufacturing/biosimilars/opportunities-challenges-biosimilar-development/.
[5] EMA authorization details[EB/OL]. (2017-03-22)[2018-2-9].
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004212/human_med_002081.jsp&mid=WC0b01ac058001d124.
[6] EMA authorization details[EB/OL]. (2017-11-10)[2018-2-9].
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004319/human_med_002181.jsp&mid=WC0b01ac058001d124.
[7] EMA authorization details[EB/OL]. (2017-8-24)[2018-2-9].
http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004279/human_med_002147.jsp&mid=WC0b01ac058001d124.
[8] EMA authorization details[EB/OL]. (2019-8-2)[2019-1-9].
https://www.ema.europa.eu/en/medicines/human/EPAR/hyrimoz#product-information-section
[9]U.S. Food and Drug Administration. approval letter[EB/OL]. (2016-9-23)[2018-2-9].
https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2016/761024Orig1s000ltr.pdf.
[10]U.S. Food and Drug Administration. approval letter[EB/OL]. (2017-8-25)[2018-2-9].
https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761058Orig1s000ltr.pdf.
[11] U.S. Food and Drug Administration. approval letter[EB/OL]. (2018-10-20)[2019-1-9].
https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/761071Orig1s000ltredt.pdf.
[12]国家食品药品监督管理总局.生物类似药研发与评价技术指导原则(试行) [EB/OL].(2015-02-28)[2018-2-9].
http://www.sda.gov.cn/WS01/CL0087/115103.html.
[13] 国家食品药品监督管理总局. 以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则[EB/OL]. (2016-03-18)[2018-2-9].
http://www.sda.gov.cn/WS01/CL0087/147583.html
[14] 中华医学会风湿病学分会. 2018中国类风湿关节炎诊疗指南[J]. 中华内科杂志, 2018(4).
[15] U.S. Food and Drug Administration. FDA Presentations for the August 3,2017 Meeting of the Arthritis Advisory Commtittee[EB/OL]. [2018-7-20].
https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM511899.pdf.
[16] European Medicines Agency. Imraldi: EPAR-public assessment report[EB/OL]. (2017-8-31) [2018-7-20].
https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM511899.pdf.
[17] Van d H D, Kivitz A, Schiff M H, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double-blind, placebo-controlled trial[J]. Arthritis & Rheumatism, 2006, 54(7):2136-2146.
[18]Huang F, Gu J, Zhu P, et al. Efficacy and safety of adalimumab in Chinese adults with active ankylosing spondylitis: results of a randomised, controlled trial.[J]. Annals of the Rheumatic Diseases, 2014, 73(3):587-94.
[19]U.S. Food and Drug Administration. Biosimilar Product Regulatory Review and Approval[EB/OL]. https://www.fda.gov/downloads/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/ucm581309.pdf.
[20] Tesser J R, Furst D E, Jacobs I. Biosimilars and the extrapolation of indications for inflammatory conditions[J]. Biologics Targets & Therapy, 2017, 11:5.
表1 修美乐在美国、欧盟和国内批准的适应症


来源:中国药审 作者:CDE

为你推荐
 资讯
资讯 国家药监局批准两款创新医疗器械
近日,国家药监局批准两款创新医疗器械上市。分别为苏州无双医疗设备有限公司的植入式心律转复除颤器和深圳大医伽玛刀科技有限公司的头部伽玛射线立体定向放射治疗系统。
2026-07-15 18:34
 资讯
资讯 翰森制药最高23亿美元授权并参与纳斯达克上市公司股权投资
7月14日,翰森制药发布公告宣布,已与Avere Therapeutics, Inc (Avere)签订独家许可协议及参与战略投资。
2026-07-15 14:20
 资讯
资讯 2026年底前至少完成一轮集中采购,国家卫生健康委 国家中医药管理局 国家疾病预防控制局关于进一步做好公立医疗卫生机构医用设备集中采购工作
国家级集中采购。国家卫生健康委继续负责组织全国公立医疗卫生机构甲类大型医用设备集中采购。个性化定制程度高、需配套设计并建设基础设施工程、逐台按程序获得注册的设备,待...
2026-07-15 09:27
 资讯
资讯 中国医药工业主营业务收入100强
近日,由中国医药工业信息中心举办的2026第43届全国医药工业信息年会在北京开幕,备受关注的“2025年度中国医药工业主营业务收入前100位企业”同步揭晓。
2026-07-14 19:55
 资讯
资讯 国家药监局批准1类创新药阿更葡糖钠注射液上市
近日,国家药品监督管理局批准杭州奥默医药股份有限公司申报的1类创新药阿更葡糖钠注射液(商品名称:奥美克松)上市,该药品用于拮抗罗库溴铵诱导的神经肌肉阻滞。该药品上市为...
2026-07-14 17:01
 资讯
资讯 迪哲医药最高15亿美元BD阿斯利康
今日,迪哲医药发布公告,公司授予阿斯利康在全球范围内的独家开发、商业化舒沃哲的权利。公司将获得阿斯利康支付的一次性、不可返还的首付款6亿美元,最高达4亿美元的临床开发...
2026-07-14 14:57
 资讯
资讯 国家医保局、国家药监局开展跨部门联合检查
近日,国家医保局、国家药监局组成联合检查组,依据药品追溯码筛查线索对内蒙古自治区和山西省部分地市医药机构、药品批发企业开展联合检查。国家医保局党组成员、副局长黄华...
2026-07-14 11:46
 资讯
资讯 2026年国家医保目录及商保目录调整通过形式审查名单公布
2026年目录调整申报阶段,国家医保局共收到基本医保目录申报信息800份,涉及药品通用名664个,最终601个通过形式审查,其中目录外368个,目录内233个,总体通过率91%。共收到商...
2026-07-14 10:34


 资讯
资讯 全国首笔药品追溯码无追索权保理业务在温州落地
近日,在温州市医保局的积极推动下,全国首笔基于药品追溯码的无追索权保理业务成功落地。国药控股温州有限公司凭借药品追溯码形成的真实交易记录,将其对温州医科大学附属眼视...
2026-07-13 10:15

 资讯
资讯 时隔8年调整的2026版国家基药目录新增的4种1类创新药和独家品种
7月9日,国家卫生健康委、国家中医药局、国家疾控局三部门公布2026年版国家基本药物目录。国家基本药物目录在新医改启动后,于2009、2012、2018年完成三轮更新,这是时隔8年后的...
2026-07-12 21:31
 资讯
资讯 陈薇院士出任中国工程院党组成员、副院长
据中国工程院官网消息,根据中共中央和国务院通知,中国工程院第九届院领导班子组成如下:张玉卓同志任党组书记、院长;张军、陈杰、李仲平、陈建峰、陈薇等同志任党组成员、副...
2026-07-11 21:36

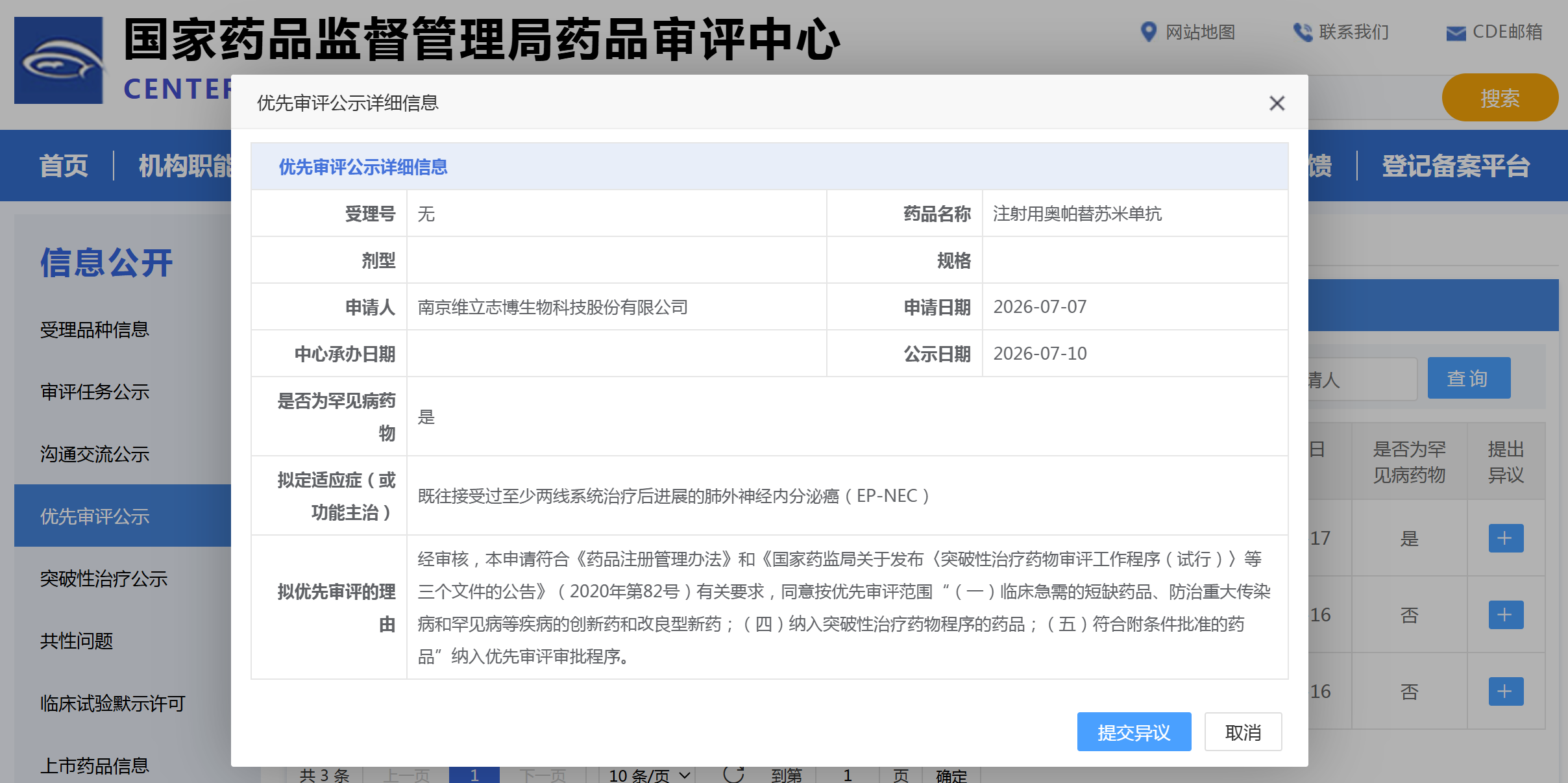 资讯
资讯 维立志博PD-L1/4-1BB 双抗拟纳入优先审评
7月10日,国家药监局药品审评中心官网显示,南京维立志博生物自主研发的注射用奥帕替苏米单抗拟纳入优先审评品种,申报适应症为既往接受过至少两线系统治疗后进展的肺外神经内分...
2026-07-10 15:08
 资讯
资讯 丰原药业实控人拟变更为安徽蚌埠市国资委
7月9日,丰原药业发布公告,公司控股股东安徽丰原集团有限公司的一致行动人7月8日与蚌埠投资集团有限公司签署《股份转让意向协议》,蚌埠投资集团拟受让丰原集团的一致行动人持...
2026-07-10 13:49