
7月11日,国家食品药品监督管理总局药品审评中心发布《关于“新注册分类的皮肤外用仿制药的技术评价要求”公开征求意见的通知》。
通知称,局部作用、局部起效的皮肤外用制剂,是药物分类与管理中的重要分支,主要分为处方药和非处方药(OTC) 两大类。2016年3月原国家食品药品监督管理总局在《关于发布化学药品注册分类改革工作方案的公告》中明确提出,按新注册分类申报的注册分类4的仿制药应与原研药的质量与疗效达到一致。
为落实该要求,并推动我国化学仿制药的注册技术标准与国际接轨,促进仿制药研发和生产水平的提升,该中心结合国际相关技术指南以及我国仿制药研发与审评的现状,通过文献调研、多专业的合审会、主任专题会议以及专家咨询会等途径,对新注册分类的皮肤外用仿制药的技术要求进行了深入探讨,形成了初步的技术要求草案并向社会各界征求意见,请8月12日前回馈意见。
联系人:孙元朋
电话: 8524-2656
邮箱: sunyp@cde.org.cn
医谷+
新注册分类下皮肤科仿制药的技术评价要求(征求意见稿)
一、本技术要求仅针对新注册分类4及5.2类的局部作用、局部起效的皮肤科外用制剂。对施用于局部,发挥全身或系统性疗效的皮肤外用药品,应按相关适应症的技术要求予以评价,不在本技术要求范围之内。
二、申请人应全面了解已上市皮肤科外用药品的国内外上市背景、安全性和有效性数据、上市后不良反应监测情况,评价和确认其临床价值。
三、已上市皮肤科外用药品具有完整和充分的安全性、有效性数据的,或被FDA橙皮书收载的,按本技术要求选择参比制剂,参照本技术要求和国内外相关技术指导原则开展研发工作。
四、已上市皮肤科外用药品不具备完整和充分的安全性、有效性数据的,应按照新药的技术要求进行研发,以确认其安全性、有效性和质量可控性。
五、参比制剂的选择
作为参比制剂的原研产品应具有完整和充分的安全性、有效性数据作为上市依据,符合以上要求的,建议申请人按以下顺序选择参比制剂:
(一)首选国内上市的原研药;如原研药国内尚未上市,应选择在美欧日已上市的原研药品。如原研药品在不同国家的上市处方不一致,申请人可按照现行技术要求进行评估,选择更合理的原研药品。
(二)在原研厂停止生产的情况下,可选择美欧日获准上市并获得参比制剂地位的药品。
六、处方工艺技术要求
(一)剂型及处方
1、一般认为,仿制药与参比制剂的辅料种类及用量(Q1和Q2)的一致,会有助于产品质量与疗效一致性的评价。故建议申请人通过查阅参比制剂说明书、专利、文献或适当的处方解析手段(如逆向工程等),对参比制剂处方进行解析,并在此基础上对处方进行合理的开发,以保证仿制品与参比制剂原辅料的种类及用量尽可能一致。辅料的用量相同是指仿制药辅料用量为参比制剂相应辅料用量的95%-105%。
2、软膏剂、凝胶剂:该类制剂采用均相基质,需注意分析与参比制剂基质类型及种类的差异性,原则上不允许改变基质的种类。
乳膏剂、乳剂:该类制剂基质一般由油相和水相组成,需关注与参比制剂的处方差异。原则上仿制药应选用与原研产品类型一致的表面活性剂。工艺方面应注意分析工艺条件及关键工艺参数的合理性,明确与参比制剂工艺的差异。
对主药呈混悬状态的局部外用制剂,应对主药的粒径与粒径分布等指标加以控制,并通过体外溶出研究等手段来确认与原研产品释放行为的一致性。
3、需阐明处方中抑菌剂、稳定剂和抗氧剂的加入理由,提供其对申报品的安全性和有效性的影响研究资料或文献资料,并在成品标准中建立合适方法加以控制。
4、需注意关注辅料对皮肤透过作用的影响,如基质特性、亲脂性溶媒、表面活性剂对皮肤角质层细胞通透性的影响等。
对于处方中添加了透皮吸收促进剂的产品,应着重考察透皮吸收促进剂的选择依据、用量筛选,并提供相应的安全性依据。必要时结合临床评价其添加的合理性和必要性。
5、辅料的浓度或用量需符合FDA IID数据库限度要求,或提供充分依据。
6、过量投料(overage):建议参考ICH Q8相关要求。
(二)生产工艺
1、工艺研究
软膏剂、凝胶剂,需注意对原料的预处理工艺(如微粉化处理)、加入方式及分散手段进行研究。需保证仿制药与参比制剂中药物晶型、粒度及粒度分布、含量均匀性等关键质量指标的一致。
乳膏剂、乳剂,需对物料加入的顺序、溶解温度、剪切速度及时间进行研究,关键工艺参数可能对产品质量产生较大影响,进而影响到产品的有效性。
2、工艺验证
(1)工艺验证内容
应至少包含:
设备的选择和评估;
工艺步骤合理性的评估;
工艺条件/工艺参数及工艺参数的可接受范围的确认;
关键工艺步骤及工艺参数可接受的操作范围的确认;
中间体产品、成品质控项目、分析方法及质控限度的合理性分析。
(2)提交资料
应至少提供工艺验证方案和批生产记录样稿,同时提交上市后对前三批商业生产批进行验证的承诺书。
3、批量
注册批样品应在商业化生产线上生产,批量应符合以下要求:
外用溶液剂:3批注册批样品中至少有2批满足以下要求:(1)拟申报最大商业批量的10%,或(2)对于装量为2ml以上的,批量至少为50L,对于装量为2ml及2ml以下的,批量至少为30L。如同时符合(1)和(2),应选择其中批量更大的。第3批样品批量不得低于注册批最大批量的25%。
乳膏剂/软膏剂/凝胶剂:注册批三批均应至少达到100kg/批或者拟定商业化生产规模的10%(两者中选更多的)。
对于特殊原因(商业因素除外)无法满足上述基本要求的,建议在申报前与监管机构进行沟通。
七、原辅包质量控制技术要求
(一)原料药
制剂生产商需结合原料药生产工艺,根据现有指导原则和相关文件(含国家局2008年7号文)、国内外药典标准,对原料药的质量进行充分研究与评估,原则上应不低于国内外现行版药典标准。
对于原料药以混悬形式存在于制剂产品中的药物,应对其晶型、粒度分布等加以研究及控制。
(二)辅料
辅料应符合现行版中国药典要求。中国药典未收载的辅料可按美、欧、日等药典标准加以要求。国内外药典均未收载的外用药辅料,可以参考化妆品、食品标准制定相应的符合当前版中国药典要求的药用内控标准。
对某些大分子聚合物等关键性辅料,应结合其修饰基团种类、数量、聚合度、分子量分布等特性指标加以控制,同时对批次、供应商等可能会影响质量的因素也应予以关注。
(三)包材
直接接触药品的包装材料和容器应符合总局颁布的包材标准。
根据产品特点选择合适的包装材料,并根据影响因素试验、加速试验和长期试验研究结果确定所采用的包装材料和容器的合理性。
原则上,所选择内包材对产品质量保护作用,应不得低于参比制剂所用包材。
注意根据产品特性和临床需求选择合理的包材尺寸。
(四)原辅包的关联审评
制剂生产商所用的原料药、辅料及包材,应当按照国家药监局关于原料药、辅料及包材审评审批有关事项方面的公告要求,在登记平台进行相关资料的提交,并获取登记号。
八、质量研究与控制技术要求
(一)应根据产品特性和相关技术指导原则科学设计试验,提供充分的试验资料与文献资料。
(二)根据目标产品的质量概况(QTPP)确立制剂的关键质量属性(CQA)。
皮肤外用制剂的CQA一般包括但不限于以下研究:(至少三批受试制剂和参比制剂的)外观、混悬药物的多晶型形式、显微分析液滴粒径(globule size)、流变特性、pH值、黏度/锥入度、粒度、含量均匀度、微生物限度、有关物质、抑菌剂含量及抗氧剂含量、无菌(用于烧伤(除轻度I°或II°外)或严重创伤的无菌制剂)等,以及外用药的体外释放试验和/或体外透皮试验(受试制剂和参比制剂均至少一批样品)的对比研究资料。
1、晶型
原料药的晶型影响到药物的溶解速度,对制剂的制备、释放和稳定性均有着显著的影响。应对仿制品的晶型、制备过程和稳定性研究中的转晶现象加以深入研究,以阐明晶型可能对药物的安全性和有效性的影响。
2、粒度分布和液滴粒径(globule size)
药物在混悬状态下,粒径及其分布对药物的溶解度、释放速率可能会有较大的影响。建议按中国药典四部通则0109项下的要求,采用包括显微镜、电镜等合适手段,与参比制剂的粒度大小及分布进行比较,并在稳定性研究中考察粒度的变化情况。
乳膏剂、乳剂产品为包含油/水两相的热力学不稳定体系,制剂的液滴粒径(globule size)和流变学特性等指标反映了处方工艺的合理性,并可能会影响到药物的释放效果和临床疗效。建议对仿制品与参比制剂的液滴粒径和流变特性进行全面的对比研究(剪切应力与剪切速率的完整流动曲线,确定出低或高剪切应力或速率;如测试物料表现出塑性流动行为,则应报告屈服应力值;检测并报告线性粘弹性响应),并在稳定性研究中考察液滴粒径的变化趋势。
3、黏度或锥入度
按中国药典通则要求,应对仿制品与参比制剂的黏度或锥入度进行对比研究。
4、体外释放试验
外用药物在体外局部释放的程度和速度是制剂性能的综合体现,包括原料药的溶解度、粒径以及半固体制剂的流变性等,主要用于外用制剂的药学质量控制,也可用于药品开发过程中处方工艺的筛选研究。
体外释放试验设计可参见附件1,应提供体外释放试验方法的系统的研究及验证资料。在此基础上,对仿制药与参比制剂进行体外释放度对比研究。
5、体外透皮试验
体外透皮试验,其设计初衷是为了模拟外用药物在生理条件下的透皮过程,以部分地反映外用制剂的质量与临床治疗的有效性。与体外释放试验的主要差异在于测定装置中用人体或动物的离体皮肤替代了人工膜。
体外渗透试验设计可参见附件2,应提供体外渗透试验方法的系统的研究及验证资料。在其基础上,对仿制药与参比制剂进行皮肤透过率的对比研究。
6、在体透皮试验
在体透皮试验,是采用特定的研究方法,动态地测量皮肤给药后一定时间内药物透过皮肤的速度和药物的量。该试验可以测定药物透过皮肤的真实生理效果。经调研,目前该研究在国内外均处于探索性阶段,难以形成制药工业界普遍接受的规范方法和可接受评价标准。申请人可根据需要,尝试性地开展前瞻性研究,以佐证药物的临床疗效。
7、有关物质
应根据产品的质量特点,按照相关技术指导原则以及国内外药典的收载情况,科学合理的选择有关物质的检查方法,并提供规范的方法学验证资料。并按相关技术指导原则的要求,参考参比制剂的实测结果和国内外药典收载的杂质指标,制定合理的有关物质限度。
降解产物:应重点对主药降解途径和降解产物进行研究,包括原料药与辅料和/或内包材的反应产物。原料药的工艺杂质一般不需在制剂中进行监测或说明。
异构体:对于存在立体异构体和手性异构体等情况,应根据主药的降解途径和生产工艺等研究积累异构体的变化数据,确定是否需订入标准加以控制。
遗传毒性杂质:应结合药物的吸收途径,根据相关文献、参比制剂的情况,通过对生产工艺、产品降解途径的分析,判断潜在的遗传毒性杂质对药品安全性的影响。必要时需提供有针对性的研究资料,并根据研究结果参考相关技术指导原则加以合理控制。
7、无菌要求
对临床上有无菌需求的外用制剂,应结合临床对无菌保障程度级别的要求,在原辅料控制标准、无菌生产工艺、与药品直接接触的设备及组件、包材密封容器的灭菌验证等方面进行专门的控制和设计,并提供完整的验证资料。在质量标准中应按中国药典四部通则0109【无菌】项下要求加以控制。
(三)仿制品与参比制剂应进行全面的质量对比研究(包括黏度、药物晶型、粒度及粒度分布、液滴粒径、含量均匀度、有关物质等关键质量指标),并提供皮肤外用药物的体外释放对比试验和体外透皮吸收对比试验。
参比制剂原则上应提供多批次样品的考察数据,考察与质量和疗效一致性评价紧密相关的关键质量属性。
九、稳定性研究的技术要求
外用药的稳定性研究一般包括影响因素试验、加速试验和长期试验和使用中的稳定性试验,必要时应考察中间条件下的稳定性。对采用半渗透性容器包装的制剂,应根据药典稳定性指导原则要求,采用低湿度条件进行稳定性考察。对在低温下制剂形态可能会发生变化的产品(如乳膏剂、乳剂等),建议进行低温和冻融试验。
对于内包材无法起到完全遮光保护的品种,应按照《化学药物(原料药和制剂)稳定性研究技术指导原则》的要求进行光照稳定性研究。
对于药物以混悬形式存在的软膏剂、凝胶剂等半固体制剂,建议考察稳定性过程中晶型、粒度及粒度分布的变化;对多相热力学不稳定体系的乳膏剂、乳剂,建议考察稳定性过程中液滴大小的变化和融合情况;建议在稳定性性末期进行与参比制剂的释放度对比研究。如处方中含有抗氧剂、抑菌剂等辅料,在稳定性研究中还应定量地考察这些辅料的变化情况。
十、等效性评价方法
1、外用溶液剂:仿制药与参比制剂的辅料在定性(Q1)和定量(Q2)上应相同,辅料的浓度差异不应超过±5%。关键质量属性一致的情况下,可不要求进行人体等效性试验;若仿制药与参比制剂辅料不一致,申请人需明确上述差异并证明该差异不会对药物的安全性和有效性产生影响。对有可能影响外用溶液剂渗透性的药品配方变更,如渗透促进剂的种类变更和用量增加,需酌情开展体外和体内的对比研究。
2、半固体制剂(如乳膏剂、软膏剂、凝胶剂等):在主要质量特性(黏度、药物晶型、粒度及粒度分布、液滴粒径、含量和均匀性、有关物质)一致的基础上,可通过体外释放对比(附件1)和透皮吸收对比试验(附件2)来评价仿制药和参比制剂的质量和疗效的一致性。如二者体外释放及体外透皮吸收均一致,可不要求进行非临床研究和临床试验;如药学研究难以充分评价质量一致且有合适的病理模型,可开展相应的非临床对比研究;如以上研究仍不能确定仿制药与参比制剂的生物等效,则建议开展临床对比研究。
鉴于皮肤科外用药物种类繁多,剂型多样,申请人可按如下细则进行考虑:
①从药物本身性质(分子量、油水分配系数、解离常数等)、剂型及处方、适应症及在人体的用药部位(不同适应症可做区分处理;不同的作用部位,药物要穿透的皮肤屏障是不一样的。应结合外用药物的作用部位 ,合理选择相应的评价手段(具体见②③))等综合考虑产品的等效性评价方式。
②对仅作用皮肤角质层外、难以透过皮肤屏障的药物,可在药学主要质量特性及制剂微观结构一致的基础上,开展体外释放对比研究进行评价。如仿制药和参比制剂的体外释放行为一致,可不要求进行非临床研究和临床试验。
③对作用部位在角质层内的外用制剂,在药学主要质量特性及制剂微观结构一致的基础上,可通过体外释放和体外透皮吸收对比试验来评价仿制药和参比制剂的疗效的一致性。如体外释放及体外透皮吸收一致,可不要求进行非临床研究和临床试验。
④对药物的作用部位在角质层内或角质层下方或两者兼具,药效或者作用部位浓度与药物代谢动力学呈良好相关性时,可进行与参比制剂的人体PK试验。
⑤处方中有新增的渗透促进成分,建议进行临床终点的生物等效性研究。
⑥对于含皮质类固醇激素的制剂,可选用经初筛合格的健康受试者进行人体皮肤变白试验,以替代在体皮肤吸收试验及以临床指标为终点的生物等效性研究。
3、对于局部作用的药物可能会导致全身暴露(重点关注大面积使用的局部外用制剂),而且存在一定的全身不良反应风险时(如皮质类固醇激素类产品等),建议与参比制剂进行人体PK对比试验研究。
十一、药品说明书的拟定
申请人需检索并追踪参比制剂说明书变更情况,参照参比制剂最新版说明书,拟定新注册分类的皮肤外用药品的说明书。
参考文献:
1、ICH Steering Committee, Harmonised Tripartite Guideline Q8: Pharmaceutical Development. August, 2009
2、ICH Steering Committee. Harmonised Tripartite Guideline Q1A: Stability Testing of New Drug Substances and Products. 2003
3、ICH Steering Committee. Harmonised Tripartite Guideline M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. 2017
4、《化学药物制剂研究基本技术指导原则》,2005.3.
5、 中国药典2015年版四部通则0109 软膏剂、乳膏剂
6、谢松梅,杨志敏,邵颖,等。新法规下皮肤外用药物临床试验要求。CFDA-CDE, 2009.12.
7、CDE:常用药用辅料数据库( http://www.cde.org.cn/drugInfo. do?method=init&frameStr=1 <http://www.cde.org.cn/drugInfo.%20do?method=init&frameStr=1>)
8、国家食品药品监督管理局:总局关于发布化学药品新注册分类申报资料要求(试行)的通告(2016年 第80号)
9、国家食品药品监督管理局:总局关于发布药包材药用辅料申报资料要求(试行)的通告(2016年第155号)
10、国家食品药品监督管理局:总局关于药包材药用辅料与药品关联审评审批有关事项的公告(2016年第134号)
11、国家食品药品监督管理局:总局关于调整原料药、药用辅料和药包材审评审批事项的公告(2017年第146号)
12、国家食品药品监督管理总局。《化学药物(原料药和制剂)稳定性研究技术指导原则》(2015年02月05日颁布)
13、美国FDA《联邦规章典集》,21CFR§314.94.
14 FDA: Guidance For Industry: ANDA Submissions-Refuse-to-Receive Standards (RTR Standards guidance)。 December 2016
15、FDA: Guidance for Industry: Controlled Correspondence Related to Generic Drug Development. November 2017
16、FDA: ANDA Submissions-Refuse-to-Receive for lack of Justification of Impurity Limits Guidance For Industry. August 2016
17、美国药典(USP40):通则<232>
18、美国药典(USP40):通则<3>
19、美国药典(USP40):通则<771>
20、FDA: Guidance for Industry: ANDAs Stability Testing of Drug Substances and Products. June 2013
21、FDA: Guidance for Industry: ANDAs: Stability Testing of Drug Substances and Products -Questions and Answers. May 2004
22、FDA: SUPAC-SS: Nonsterile Semisolid Dosage Forms; Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Release Testing and In Vivo Bioequivalence Documentation. May 1997.
23、FDA: Product-Specific Guidances for Generic Drug Development (https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm075214.htm)
24、 EMEA, CHMP: GUIDELINE ON EXCIPIENTS IN THE DOSSIER FOR APPLICATION FOR MARKETING AUTHORISATION OF A MEDICINAL PRODUCT. January 2008
25、EMEA, CHMP: Note for guidance on inclusion of antioxidants and antimicrobial preservatives in medicinal products. January 1998
26、 EMA, CHMP: Note for guidance on in use stability testing of human medicinal products. September 2001
27、EMA, CHMP: Concept paper on the development of a guideline on quality and equivalence of topical products. July 2015
28、PMDA:局所皮膚適用製剤(半固形製剤及び貼付剤)の処方変更のための生物学的同等性試験ガイドラインについて。2010年11月
29、PMDA: 局所皮膚適用製剤(半固形製剤及び貼付剤)の処方変更のための生物学的同等性試験ガイドラインQ&A。2010年11月
30、PMDA:局所皮膚適用製剤の後発医薬品のための生物学的同等性試験ガイドライン
31、PMDA:局所皮膚適用製剤の後発医薬品のための生物学的同等性試験ガイドラインQ&A
32、PMDA:局所皮膚適用製剤の剤形追加のための生物学的同等性試験ガイドライン。2006年11月
33、PMDA:局所皮膚適用製剤の剤形追加のための生物学的同等性試験ガイドラインQ&A。2006年11月
来源:国家药品审评中心

为你推荐
 资讯
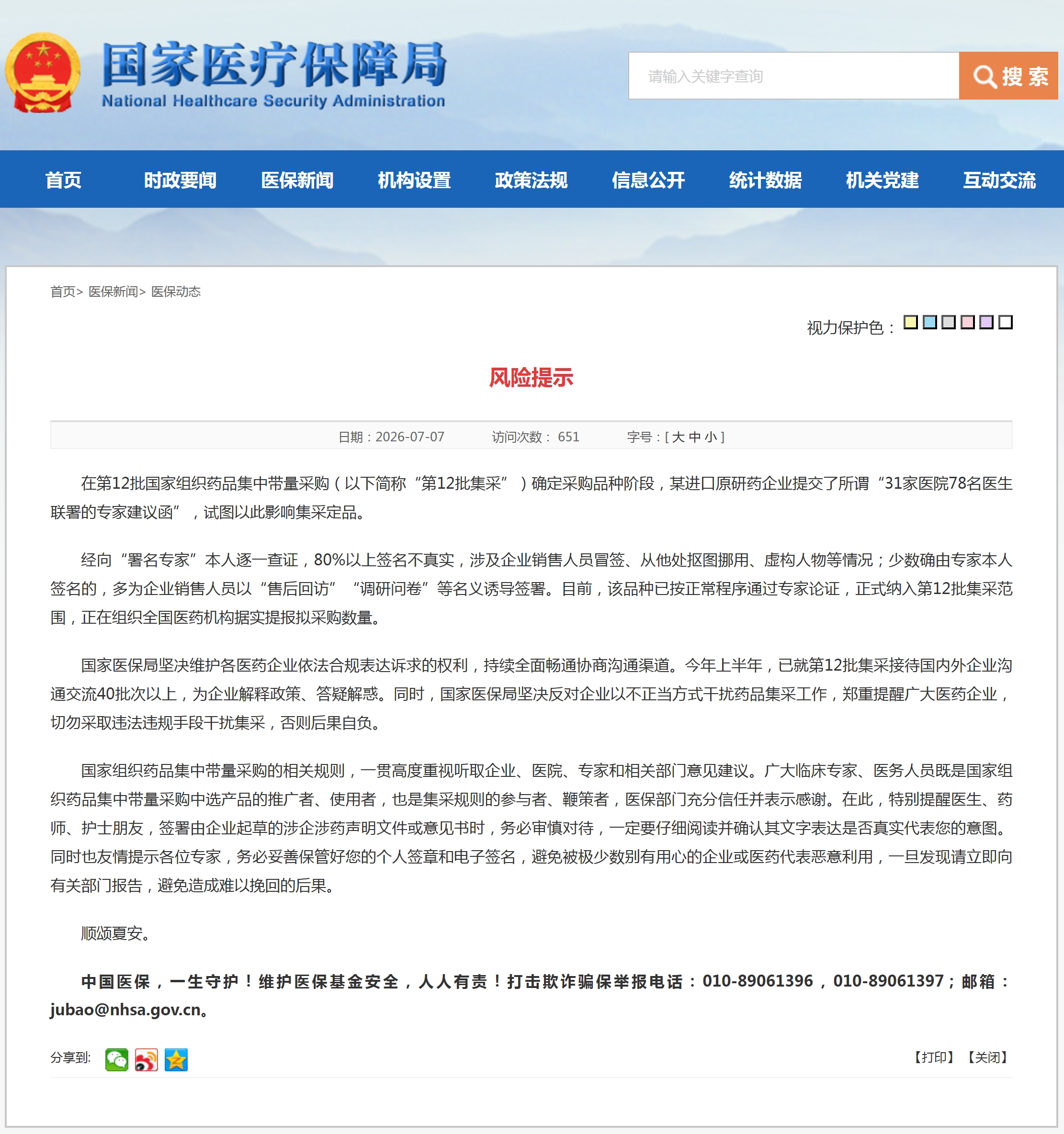
资讯 某进口原研药企提交虚假医生联署专家建议函,试图影响第12批国采
7月7日,国家医保局发布风险提示。在第12批国家组织药品集中带量采购确定采购品种阶段,某进口原研药企业提交所谓“31家医院78名医生联署的专家建议函”,试图以此影响集采定品。
2026-07-07 20:31
 资讯
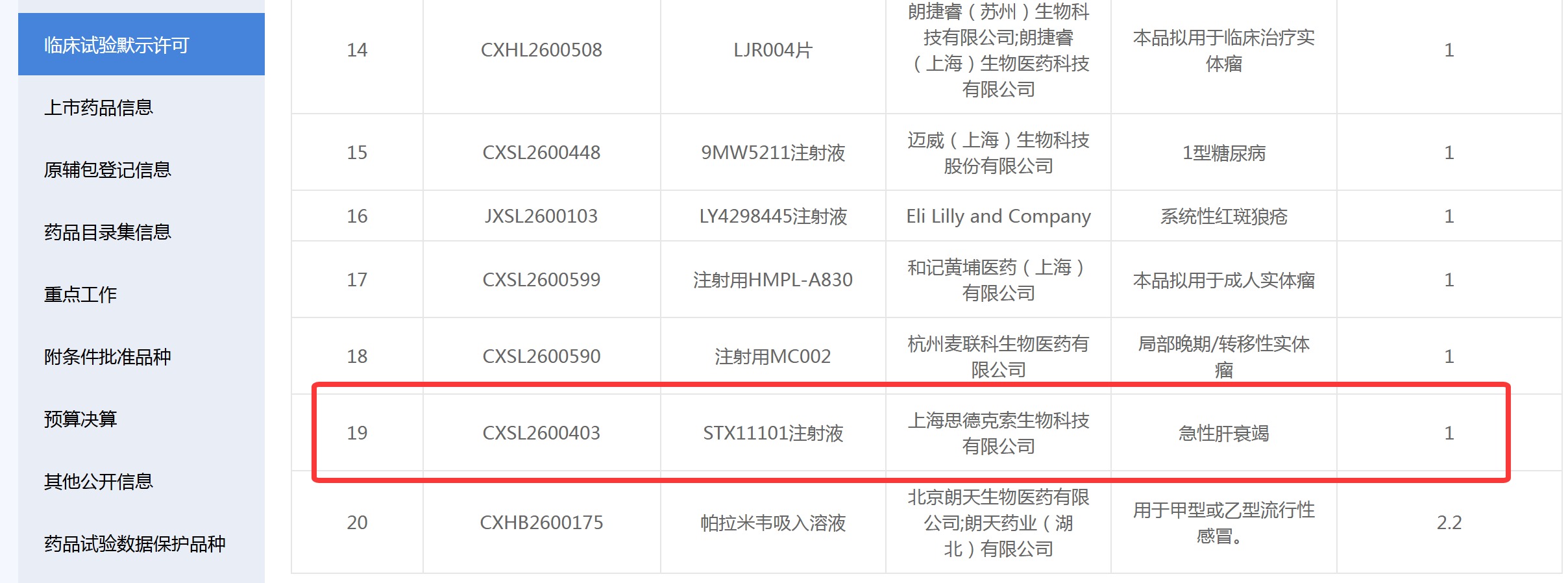
资讯 国内首款干细胞外泌体在研药物获批IND
7月6日,上海思德克索生物科技有限公司宣布,其自主研发的人源脂肪间充质干细胞来源的细胞外囊泡药物STX11101注射液,已正式获得中国国家药品监督管理局(NMPA)药品审评中心(C...
2026-07-07 15:22

 资讯
资讯 AI虚拟细胞企业华源智因获数千万元种子轮投资,水木创投注资
本轮资金将重点用于核心技术迭代、高端人才团队扩充、临床场景落地及商业化体系搭建,助力企业深耕AI计算生物学与智能药物研发领域。
2026-07-07 15:08
 资讯
资讯 中国国家药品监督管理局受理备思复(维恩妥尤单抗)联合帕博利珠单抗用于肌层浸润性膀胱癌的上市许可申请
用于肌层浸润性膀胱癌(MIBC)成人患者的新辅助治疗(术前治疗)及在膀胱切除术(手术)后的辅助治疗(术后治疗),无论患者是否耐受含顺铂化疗。
2026-07-07 14:52
 资讯
资讯 多方共话青少年成长与健康认知,探索亲子沟通与科学预防路径
近日,围绕青少年成长过程中的亲子沟通与健康认知议题,由《嘉人Marie Claire》杂志主办、默沙东中国支持的“默默爱着你”主题论坛于北京顺利举办
2026-07-07 14:49
 资讯
资讯 同仁堂今日港交所上市,破发
今日,同仁堂医养正式登陆港交所,发售价为5 5港元 股。截至7月7日中午收盘,同仁堂医养下跌超过40%,市值不足15亿港元。另值得注意的是,公司开盘报4 76港元 股,较5 5港...
2026-07-07 13:35
 资讯
资讯 歌礼制药两款丙肝治疗药物正式调出医保目录
由于2025年医保谈判中没有成功续约,在6个月的过渡期后,日前,歌礼制药的达诺瑞韦钠片(戈诺卫)、盐酸拉维达韦片(新力莱)被正式调出医保目录。
2026-07-07 11:14

 资讯
资讯 诺华15亿美元收购Myricx Bio,拥有N-肉豆蔻酰转移酶(NMT)独特药物开发机制
2026年7月6日,诺华宣布与总部位于英国的生物技术企业 Myricx Bio达成收购协议,交易总金额最高可达 15 亿美元,其中 11 亿美元为前期现金对价,剩余为潜在里程碑付款。
2026-07-06 14:17
 资讯
资讯 1998年-2025年全国经济人口卫生医保相关数据
2016年、2017年不含未整合的新型农村合作医疗参保人数,2018年起居民医保数据含整合后的新型农村合作医疗参保人数。
2026-07-06 11:16
 资讯
资讯 海正药业:获产品独家授权许可协议,首付款4500万元
近日,海正药业发布公告,公司与艾缇亚(上海)制药有限公司签署产品独家授权许可及合作协议,引进由艾缇亚开发的化药1类创新药TISA-818系统给药制剂在合作区域(中国大陆、香港...
2026-07-06 11:04
 资讯
资讯 IBD公益短剧《我和我们》首映:23集真实故事,看见被疾病遮蔽的人生
7月3日,全网首部聚焦炎症性肠病(IBD)患者群体的公益短剧《我和我们》在上海国泰电影院首映。该剧历时一年多打磨,以23集短剧体量,基于真实患者经历创作,探索疾病科普与公益...
2026-07-05 19:42
 资讯
资讯 又一家上市药企实控人涉案
6月24日,华恒生物(688639 SH)收到公司实际控制人、董事长、总经理郭恒华女士家属告知,其收到合肥市公安局蜀山分局的拘留通知书,公司实际控制人、董事长兼总经理郭恒华女士...
2026-07-05 13:24
 资讯
资讯 国家药监局:将符合条件的细胞与基因治疗药品纳入创新药临床试验审评审批30日通道
国家药监局综合司公开征求《关于优化细胞与基因治疗药品审评审批有关事项的公告(征求意见稿)》意见。
2026-07-04 10:22
 资讯
资讯 虚开价税合计金额1.89亿元,沈阳奥吉娜被取消挂网
近日,辽宁省公共资源交易中心发布《沈阳奥吉娜药业有限公司医药价格和招采信用评价结果》,沈阳奥吉娜失信等级被评定为“特别严重失信”,失信行为是存在让他人为自己虚开增值...
2026-07-03 15:14
 资讯
资讯 石药创新与阿斯利康达成新型小干扰核酸候选药物BD合作,里程碑付款最高17.4亿美元
7月2日,晚间,石药创新发布公告,公司控股子公司石药集团巨石生物制药有限公司与阿斯利康于 2026 年 7 月 1 日签署《合作、选择权及授权协议》,将与阿斯利康在新型小干...
2026-07-03 11:42
